
Fotosynteza - jest to biochemiczny proces wytwarzania związków organicznych z materii nieorganicznej, przez komórki zawierające chlorofil lub bakteriochlorofil, przy udziale światła.
1) BARWNIKI FOTOSYNTETYCZNE, BARWNIKI ASYMILACYJNE - jest to grupa organicznych związków chemicznych, najczęściej pochodzenia lipidowego. Występują w specjalnych układach, głównie w komórkach zdolnych do przeprowadzania fotosyntezy. Odgrywają kluczową rolę w tym procesie - pochłaniają energię świetlną o określonej długości fali w celu zmagazynowania jej w postaci ATP i trioz. Określone grupy barwników nadają różną barwę komórkom lub organizmom, w których występują. Ze względu na charakter chemiczny oraz pełnioną funkcję można je podzielić na:
- tetrapirolowe barwniki podstawowe (ich głównym zadaniem jest przeprowadzanie fotosyntezy);
- chlorofile - występują w chloroplastach roślin oraz sinic (nadają zieloną barwę),
- bakteriochlorofile - jest to odmiana chlorofili, występująca u bakterii
- barwniki dodatkowe
- barwniki wspomagające:
- karotenoidy (karoteny, ksantofile) - pomagają przeprowadzać fotosyntezę, a także chronią przed działaniem promieniowania UV oraz niektórymi szkodliwymi związkami chemicznymi; występują we wszystkich organizmach fotosyntezujących i u niektórych zwierząt (nadają barwę od żółtej do pomarańczowej),
- fikobiliny - występują jedynie u niektórych glonów i sinic.
A) CHLOROFILE - jest to grupa organicznych związków chemicznych obecnych między innymi w roślinach, algach i bakteriach fotosyntezujących (np. w sinicach). Nadaje częściom roślin (głównie liściom) charakterystyczny zielony kolor.
 |
| Źródło: Wikipedia. Maksima absorpcyjne chlorofili na tle widma światła białego |
Funkcją chlorofili w organizmach przeprowadzających fotosyntezę jest wychwytywanie kwantów światła i przekazywanie energii wzbudzenia do centrum reakcji fotoukładu, skąd wybijane są elektrony, spożytkowane następnie w dalszych etapach fotosyntezy.
Znaczna zawartość chlorofili w organizmach fotosyntetyzujących jest odpowiedzialna za ich zieloną barwę. Zielony kolor chlorofilu spowodowany jest wysoką absorpcją w czerwonej i niebieskiej części spektrum światła, a niską absorpcją w zielonej części spektrum światła (długość fali 500-600 nm).
Wyróżnia się wiele rodzajów chlorofili. Najbardziej rozpowszechnione w przyrodzie to chlorofil a i chlorofil b występujące u wszystkich roślin przeprowadzających fotosyntezę. Chlorofile c i d występują jedynie u części glonów. U prokariontów zdolnych do przeprowadzania fotosyntezy mogą występować: chlorofil a (tylko u sinic) oraz wiele rodzajów bakteriochlorofili oznaczanych literami od a do g.
 |
| Źródło: Dummie.com - Rodzaje chlorofili, a podstawniki szkieletu porfirynowego. |
 |
| Źródło: pnas.org - Przemiany form chlorofili. |
 |
| Źródło: Wikipedia. Układ porfirynowy. |
BUDOWA CHLOROFILI: Cząsteczka każdego chlorofilu zbudowana jest z pochodnej porfiryny określanej feoporofiryną. Feoporofiryna to pięciopierścieniowa porfiryna z różnymi podstawnikami. Cztery z pierścieni to pierścienie pirolowe, a piąty pierścień tworzą same atomy węgla. Wiązania pomiędzy atomami tworzącymi pierścienie to następujące po sobie wiązania pojedyncze i podwójne składające się na układ wiązań sprzężonych. Centralne miejsce wukładzie porfiryny zajmuje atom magnezu łączący się z atomami azotu każdego z pierścieni. Pofiryna tworząca kompleks z magnezem posiada zdolność do absorpcji promieniowania elektromagnetycznego w zakresie widzialnym. Obecność magnezu wpływa na zdolność agregacji cząsteczek chlorofilu, co ułatwia przekazywanie energii wzbudzenia pomiędzy cząsteczkami.

U większości chlorofili (poza chlorofilami c) feoporfiryna łączy się poprzez wiązanie estrowe z alkoholem o 20 atomach węgla - fitolem. Przyłączony alkohol izoprenowy nie wpływa znacząco na zdolność absorpcji światła. Jego rolą jest tworzenie hydrofobowego fragmentu cząsteczki łączącego chlorofil z błonami białkowo-lipidowymi. W bakteriochlorofilach zamiast fitolu może występować inny alkohol farnezol lub geranylogeraniol występujący niekiedy w bakteriochlorofilu a.
Do układu porfiryny w różnych miejscach przyłączone są dodatkowe grupy. Wpływają one na niewielkie zmiany zdolności absorpcji kwantów światła przez poszczególne rodzaje chlorofili. Do układu porfiryny w różnych miejscach przyłączone są dodatkowe grupy. Wpływają one na niewielkie zmiany zdolności absorpcji kwantów światła przez poszczególne rodzaje chlorofili. W zależności od podstawników układu porfirynowego wyróżnia isę następujące typy chlorofilu:

Dwa najpowszechniej występujące chlorofile, chlorofil a - niebieskozielony, chlorofil b - żółtozielony, stanowią przeważającą większość masy wszystkich barwników w organie fotosyntetyzującym.
 |
| Źródło: Wikipedia. Chlorofil może absorbować kwanty światła czerwonego i niebieskiego. |
ABSORPCJA ŚWIATŁA I UDZIAŁ W FOTOSYNTEZIE: Maksimum absorpcji dwóch najczęściej występujących chlorofili 420 i 662 nm dla chlorofilu a oraz 453 i 642 nm dla chlorofilu b. Po raz pierwszy widmo absorpcyjne chlorofilu wyznaczył w 1883 r. niemiecki biolog Theodor Wilhelm Engelmann. Maksymalny molowy współczynnik absorpcji dla chlorofilu a wynosi 105 M−1 cm−1 i jest jednym z najwyższych wyliczonych dla związków organicznych. Cząsteczka chlorofilu po zaabsorbowaniu kwantu światła (fotonu) ulega wzbudzeniu. Pochłonięcie kwantu światła czerwonego wiąże się z przejściem do pierwszego stanu wzbudzonego, pochłonięcie kwantu światła niebieskiego skutkuje przejściem do drugiego stanu wzbudzonego. Stan wzbudzenia przekazywany jest przez kolejne cząsteczki chlorofilu do centrum reakcji - pary cząsteczek chlorofilu a w specyficznym otoczeniu białkowym. Z chlorofilu stanowiącego centrum reakcji elektron jest wybijany, dochodzi do fotoindukcyjnego rozdziału ładunków, i następnie przechwytywany przez kolejnych pośredników zlokalizowanych w obrębie fotosystemów, a następnie na kolejne przekaźniki w obrębie błony tylakoidów, biorące udział w fotosyntetycznym łańcuchu transportu elektronów. Transport elektronów w błonach tylakoidów jest konieczny do wytworzenia NADPH (tzw. "siły redukcyjnej") oraz gradientu protonowego w poprzek błony tylakoidu, co jest konieczne do produkcji ATP przez chloroplastową syntazę ATP. W chloroplastach, chlorofil wchodzi w skład większych kompleksów barwnikowo-białkowych (tak zwanych fotosystemów oraz układów antenowych). Stosunku ilościowe chlorofili w roślinach zależą między innymi od warunków siedliskowych: rośliny cieniolubne (cienioznośne) mają więcej chlorofilu b, rośliny światłolubne (światłożądne) - chlorofilu a.
 |
| Źródło: Wikipedia. Przylaszczka - skiofit runa leśnego. |
- Rośliny cieniolubne, skiofity (gr. skia = cień + phyton - roślina), rośliny cienioznośne - są to rośliny, które przystosowane są do życia w warunkach dużego zacienienia. Rośliny te źle rosną w pełnym słońcu i nie wytrzymują w tych warunkach konkurencji z innymi roślinami. Do roślin cieniolubnych zalicza się m.in. szczawik zajęczy (Oxalis acetosella), kopytnik pospolity (Asarum europaeum), przylaszczka pospolita (Hepatica nobilis), marzanka wonna (Galium odoratum). Dużo roślin cieniolubnych rośnie w warstwie runa leśnego, gdzie z natury jest duże zacienienie spowodowane przez korony drzew. W rzeczywistości występowanie tych roślin w środowiskach zacienionych spowodowane jest w większym stopniu wilgotnością i brakiem konkurencji, niż światłem. Skiofity mają liście z reguły duże, cienkie, bez nalotu woskowego i włosków. Liczba apratów szparkowych jest mniejsza, niż u heliofitów (roślin światłolubnych). Ich liście, a często i łodygi, są zwykle ciemnozielone, gdyż zawierają dużo chlorofilu. Fotosynteza zachodzi u nich najintensywniej przy średnim oświetleniu. W liściach miękisz gąbczasty jest luźny, tkanka przewodząca słabo rozwinięta. Niektóre rośliny są skiofilami tylko na niektórych etapach rozwojowych. Młode siewki wielu drzew leśnych (np. buka pospolitego (Fagus sylvatica), świerka pospolitego (Picea abies) są wybitnymi skiofilami, w późniejszym natomiast okresie, gdy stają się drzewem, mogą rosnąć w pełnym słońcu. W porównaniu z roślinami lądowymi skiofitami są również glony, zwłaszcza bentoniczne, gdyż woda, w której żyją rozprasza światło. Wiele glonów żyjących w głębszych warstwach toni wodnej do wychwytywania energii słonecznej używa dodatkowych barwników fotosyntetycznych, w celu wykorzystania światła niepochłanianego przez chlorofil, przez co przybiera inną niż zielona barwę.
 |
| Źródło: Wikipedia. Rojniki są typowymi heliofitami. |
- Rośliny światłolubne, heliofity, rośliny światłożądne - są to rośliny wymagające do swojego rozwoju dużej ilości światła. Rośliny te mogą się prawidłowo rozwijać tylko w środowisku o pełnym nasłonecznieniu. W zacienionych miejscach rozwijają się słabo, lub giną. Rośliny światłolubne mają specjalne przystosowania. Ich liście mają grubą skórkę, często dodatkowo pokrytą woskowym nalotem. Zazwyczaj posiadają bogate nerwację i dużą ilość aparató∑ szparkowych, co zwiększa skuteczność transpiracji zapobiegającej przegrzaniu liści. Niektóre gatunki mają liście błyszczące, wskutek czego odbijają część światła, zapobiegając w ten sposób przegrzewaniu się liści i zmniejszając transpirację. Charakterystyczną cechą roślin światłolubnych jest także duża intensywność oddychania. W liściach miękisz palisadowy jest dobrze rozwinięty, często wielowarstwowy. Do wytworzenia takiej samej ilości asymilatów, jak rośliny cieniolubne potrzebują nieraz kilkakrotnie więcej światła. Heliofitami wśród polskich drzew są np. modrzew (Larix sp.) i brzoza (Betula sp.), wśród roślin uprawnych kukurydza (Zea mays), soja zwyczajna (Soja max). Heliofitami są rośliny pustyń i rośliny wysokogórskie, gdyż rośliny te z reguły rosną w pełnym słońcu, w środowisku niezacienionym przez drzewa i krzewy. Wiele jest roślin światłolubnych wśród roślin ozdobnych. Należą do nich np. różne odmiany róży (Rosa sp). Dużej ilości światła wymagają odmiany roślin ozdobnych o liściach wybarwionych na różne odcienie koloru żółtego i czerwonego. W roślinach tych zielony barwnik chlorofil, niezbędny do fotosyntezy, przytłumiony jest przez inne barwniki, jak np. u trójskrzynu pstrego (Cordiaeum viaregatum), zwanego krotonem.
SYNTEZA CHLOROFILI: Miejscem syntezy chlorofili u roślin są plastydy. To w nich przebiegają wszystkie reakcje prowadzące do wytworzenia cząsteczki chlorofili. Początkowym substratem służącym do syntezy chlorofili jest jeden z aminokwasów białkowych - kwas glutaminowy. Pierwszym etapem jest aktywacja aminokwasu polegająca na przyłączeniu cząstki tRNAGlu przez syntazę glutamylo-tRNAGlu. Reakcja ta wymaga hydrolizy jednej cząsteczki ATP do AMP i PPi. Powstający glutamylo-tRNAGlu redukowany jest do 1-semialdehydu glutaminianowego przez reduktazę Glu-tRNA. Reakcja ta wymaga zużycia cząsteczki NADPH. Powstały 1-semialdehyd glutaminianowy przekształcany jest przez aminotransferazę semialdehydu glutaminianowego do kwasu 5-aminolewulinowego (ALA).
 |
| Źródło: Wikipedia. Synteza chlorofili.
Dwie cząsteczki tego niebiałkowego aminokwasu w reakcji kondensacji katalizowanej przez enzym dehydratazę ALA przekształcane są do porfobilinogenu (PBG). Deaminaza porfobilinogenowa odłączając reszty aminowe - NH2 łączy cztery cząsteczki porfobilinogenu w hydroksymetylobilan. W kolejnej reakcji następuje zamknięcie pierścienia przez syntazę uroporfirynogenu III. Powstający uroporfirynogen III ulega dekarboksylacji i przekształcany jest w koproporfirynogen III przez dekarboksylazę uroporfirynogenu III. Koproporfirynogen III jest utleniany przez oksydazę porfirynogenu III do protoporfirynogenu IX i przez oksydazę protoporfirynogenu IX do protoporfiryny IX. Reakcje prowadzące do powstania protoporfiryny IX zachodzą w stromie plastydów.
Kolejne etapy syntezy zachodzą na błonach osłonki plastydu w której znajdują się odpowiednie enzmy. Do pierścienia protoporfiryny IX Mg-chelataza wprowadza atom magnezu. Transferaza przyłącza resztę metylową w pozycji 15, a cyklaza zamyka piąty pierścień obecny w chlorofilu. Powstały diwinyloprotochlorofilid a redukowany jest do monowinyloprotochlorofilidu a przez reduktazę winylową zależną od NADPH. Powstały po redukcji monowinyloprotochlorofilid a redukowany jest do chlorofilidu a przez oksydoreduktazę protochlorofilidu. Reakcja ta wymaga udziału NADPH oraz światła, ponieważ redukcji może ulec jedynie monowinyloprotochlorofilid wzbudzony kwantem światła. Podczas redukcji likwidacji ulega jedno z wiązań podwójnych IV pierścienia pirolowego. Chlorofilid a łączony jest w reakcji estryfikacji z dwudziestowęglowym alkoholem izoprenowym - fitolem rpzez syntazę chlorofilową. Powstały chlorofil a może bezpośrednio służyć do absorpcji światła lub zostać przekształcony przez oksygenazę chlorofilu b do drugiego z najczęściej występujących chlorofili. Dwie ostatnie reakcje, a więc wytworzenie chlorofilu a lub chlorofilu b zachodzą w błonach tylakoidów.
Jeśli roślina nie znajduje się na świetle protochlorofilid, lipidy oraz oksydoreduktaza NADPH-protochlorofilid gromadzą się w strukturach określanych jako protylakoidy. Dopiero oświetlenie roślin (np. po wykiełkowaniu) pozwala na zakońćzenie syntezy chlorofili i przekształcenie protylakoidów w tylakoidy. Rośliny okrytonasienne, które nie mają dostępu do światła ulegają etiolacji, czyli rosną bez wykształcenia chlorofilu i chloroplastów, a w ich plastydach dochodzi do wykształcenia jedynie protylakoidów.
METODY BADANIA CHLOROFILI: Chlorofile są dobrze rozpuszczalne w rozpuszczalnikach organicznych (Aceton itp.) i tłuszczach, a praktycznie nierozpuszczalne w wodzie. Chlorofile w roztworach wykazują silną fluorescencję. Fluorescencja chlorofili in vivo zależy od stanu funkcjonalnego układu fotosyntetycznego i jest wykorzystywana do pomiarów parametrów wydajności fotosyntezy (metoda PAM, ang. Pulse Amplitude Modulated chlorophyll fluorescence).
ZASTOSOWANIE CHLOROFILU JAKO BARWNIKA: Chlorofil A jest używany jako barwnik w przemyśle spożywczym do produkcji np. zup, sosów, oliwy z oliwek, oleju sojowego, lodów oraz fermentowanych napojów mlecznych. Został uznany za nieszkodliwy w zastosowaniach spożywczych. Rzadko spotykanym działaniem niepożądanym chlorofilu jest uczulenie na światło. Jest również wykorzystywany w produktach takich jak antyperspiranty i płyny do płukania jamy ustnej.
B) KAROTENOIDY - jest to grupa organicznych związków chemicznych, węglowodory nienasycone o szczególnej budowie, żółte, czerwone, pomarańczowe i różowe barwniki roślinne, występujące w chloroplastach i chromatoforach. Karotenoidy należą do naturalnych przeciwutleniaczy. Karotenoidy należą do prekursorów witaminy A i są głównym dietetycznym źródłem tej witaminy u człowieka. W przewodzie pokarmowym powstaje retinal, który następnie jest przekształcany do retinolu. Jedną z funkcji karotenoidów jest zabezpieczanie przed reaktywnymi formami tlenu powstających podczas fotosyntezy (aktywność przeciwutleniająca).
BUDOWA KAROTENOIDÓW: Karotenoidy zbudowane są z jednostek izoprenowych, zawierających pięć atomów węgla, należą do 40-węglowych terpenoidów, czyli tetraterpenów. Karotenoidowy szkielet węglowy C40 budowany jest przez kolejne dodawanie jednostek C5. Z chemicznego punktu widzenia, charakterystyczną cechą karotenoidów jest występowanie dwóch pierścieni cykloheksylowych połączonych długim łańcuchem węglowym, w którym występuje układ szeregu sprzężonych wiązań podwójnych węgiel-węgiel. Są to więc mieszane cykliczno-liniowe polieny. Jak dotąd zidentyfikowano i opisano około 800 karotenoidów.
ROLA KAROTENOIDÓW: Związki te pełnią pomocniczą rolę w procesie fotosyntezy, ponieważ absorbują pewne zakresy promieniowania świetlnego (niebieska, fioletowa) aby następnie przekazywać energię stanu wzbudzonego na cząsteczkę chlorofilu. Pełnią również funkcję ochronną przed procesami fotooksydacji, na które narażone są głównie nienasycone kwasy tłuszczowe lipidów chloroplastowych. W liściach, ich barwa jest maskowana przez zieloną barwę barwników chlorofilowych, uwidocznia się to jesienią, kiedy chlorofile są degradowane przez enzymy: chlorofilazę, decheletazę i oksygenazę feoforbidu a, która katalizuje otwarcie pierścienia porfirynowego w feoforbidzie - ostatnim związku o barwie zielonej na szlaku rozkładu chlorofilu. Przykładem karotenoidu jest beta-karoten. Karotenoidy nadają również barwę innym częściom rośliny, np. korzeniowi marchewki. Zazwyczaj występują w komórce w zdecydowanie mniejszych stężeniach niż chlorofile. Nie rozpuszczają się w wodzie. Ich cechą jest fotolabilność - ulegają przemianom w obecności światła. Ponadto spełniają ważną rolę ochronną przed uszkodzeniem fotosystemu spowodowanym nadmiarem docierającej energii świetlnej, pochłaniając ją i powodując jej dyspresję (czyli rozproszenie) albo też przekierowując na inne procesy fizjologiczne w komórce.
PRZYKŁADY WYSTĘPOWANIA KAROTENOIDÓW U ROŚLIN: zielone części: w liściach i łodygach (wraz z chlorofilem w chromatoforach, w formie bezpostaciowej lub krystalicznej), nie zielone blaszki liści (liście etiolowane - przede wszystkim ksantofile), liście jesienne - po rozłożeniu chlorofilu pozostają karotenoidy, dając piękne zabarwienie; kwiaty, u rodzin jednoliściennych i dwuliściennych; owoce i ziarna; korzenie następujących roślin: Daucus cartota, Beta vulgaris, Brassica rapa, Ipomoea batatas, Escobedia sabrifolia, Pastinaca sativa.

|

 |
| Źródło: Wikipedia. Acyrthosiphon pisum - morfotyp czerwony. |
- Torulen - jest to organiczny związek chemiczny z grupy karotenoidów wykryty u mszyc z gatunku Acyrthosiphon pisum. Karotenoidy są syntetyzowane przez rośliny, grzyby i mikroorganizmy, jednak do czasu wykrycia torulenu u mszyc, nie znano szlaku biosyntezy tych związków u zwierząt. Mszyce z gatunku Acyrthospiphon pisum wykazują polimorfizm, czerwony lub zielony kolor. Torulen występuje u osobników czerwonych, co wpływa na możliwość ich obrony przed drapieżnikami. Analiza filogenetyczne wykazała, że geny niezbędne do przeprowadzenia syntezy torulenu mszyce nabyły w wyniku poziomego transfery genów od grzybów. U mszyc wykryto cztery kopie genów kodujących desaturazę karotenu i trzy dla syntazy fitoenu/cyklazy karotenu. Jednak tylko jeden gen desaturazy jest odpowiedzialny za syntezę torulenu. Transfer genów odbył się w dalekiej przeszłości od grzybów zbliżonych do Mucoromycotina.
- Witamina A - jest to zbiorcza nazwa organicznych związków chemicznych z grupy retinoidów (z których najważniejszy jest retinol), pełniących w organizmie funkcję niezbędnego składnika pokarmowego, rozpuszczalnej w tłuszczach witaminy. Witamina A jest jedną z najwcześniej odkrytych witamin. Skutki jej niedoboru znane były już w starożytnym Egipcie, Grecji oraz Rzymie i określane były nazwą kurza ślepota lub ślepota zmierzchowa. Chorobę tę leczono podając gotowaną lub surową wątrobę. Dopiero na przełomie XIX i XX wieku udało się ustaić związek pomiędzy nieprawidłowym odżywianiem a rozwojem kurzej ślepoty. Witamina A została odkryta w tranie w roku 1913 przez amerykańskich badaczy, Elmera McColluma i Marguerite Davis, zaś nazwę "witamina A" nadano jej w latach 20. XX wieku. Początkowo występowanie witaminy A w tranie łącznie z witaminą D sprawiało problemy w określeniu rzeczywistych właściwości każdej z tych witamin. W pożywieniu pochodzenia zwierzęcego, podstawową formą w jakiej występuje witamina A jest ester - palmitynian retinolu, który w jelicie cienim ulega deestryfikacji do alkoholu - retinolu. Inne ważne pochodne związane z aktywnością witaminy A to: retinal (aldehyd) i kwas retinowy (tretynoina). ŹRÓDŁA POKARMOWE WITAMINY A: Do najbogatszych naturalnych źródeł witaminy A dla człowieka należą: tran rybi (30 mg RAE/100 g), wątroba i podroby (2-28 mg RAE/100 g), słodkie ziemniaki (1 mg RAE/100 g), marchew (835-850 mikrogram RAE/100 g), jarmuż (500-700 mikrogram RAE/100 g), szpinak (ok. 500 mikrogram RAE/100 g), dynia (300-400 mikrogram RAE/100 g). Głównym źródłem aktywnych form witaminy A w organizmie jest spożywana z pokarmem pochodzenia roślinnego prowitamina A (głównie beta-karoten). W organizmie ludzkim enzymem odpowiedzialnym za przekształcenie beta - karotenu w retinal jest dioksygenaza beta - karotenowa. FIZJOLOGICZNE ZNACZENIE WITAMINY A: Witamina A spełnia wiele ważnych fizjologicznych funkcji. Funkcje retinolu (tylko on wykazuje pełną aktywność biologiczną) i retinalu: odgrywa istotną rolę w odbieraniu bodźców wzrokowych w siatkówce oka. Pochodna witaminy A, 11-cis retinal w pręcikach łączy się z białkiem opsyną, tworząc rodopsynę (purpurę wzrokową). Już nawet pojedynczy foton wzbudza fotoizomeryzacje 11-cis-retinalu do trans-retinalu, co prowadzi do pobudzenia; odpowiada za integralność błon komórkowych; odpowiada za prwaidłowe funkcjonowanie komórek tkanki nabłonkowej zarówno pokrywających jak wydzielniczych i czuciowych; reugluje wzrost tkanki nabłonkowej i innych komórek organizmu; utrzymuje prawidłowy stan skóry, włosów, paznokci; zapewnia normalny wzrost kości i zębów przez regulację aktywności komórek tkanki kostnej; chroni nabłonek układu oddechowego przed drobnoustrojami.

- Źródło: Wikipedia. Przejście formy retinalu 11-cis w trans.
- Kwas retinowy zaś: odpowiada za prawidłowe wytwarzanie kolagenu typu IV i fosfatazy zasadowej; różnicuje komórki: osteoblasty, keratynocyty, fibroblasty, spermatocyty i komórki pnia; działa przeciwnowotworowo: w ostrych białaczkach szpikowych hamuje proliferację komórek, wzmaga wydzielanie TNF alfa i aktywuje limfocyty.
- Karotenoidy są przeciwutleniaczami i działają na ogół umiarkowanie przeciwnowotoworowo, jednak u palaczy duże dawki syntetycznego beta-karotenu, zarówno powodują zwiększenie częstości występowania raka płuc i wyższą śmiertelność. Podobną zależność stwierdzono u zwierząt laboratoryjnych poddanych ekspozycji na dym tytoniowy. Gdy komórki organizmu potrzebują retinolu, to zostaje on uwolniony z wątroby do krążenia ogólnego Retinol we krwi jest transportowany przez białko RBP (retinol binding protein), kompleks ten dodatkowo łączy się z transtyretyną (białkiem, które uniemożliwia wydalenie tego kompleksu przez nerki). Kwas retinowy we krwi jest transportowany z albuminami. W komórce retinol łączy się z białkiem cRBP (cellular retinol binding protein), a kwas retinowy z cRABP. Związane cząsteczki retinoidowe mają swoiste receptory jądrowe RAR i RXR, zawierające miejsca wiążące DNA, zwane również miejscami odpowiedzi kwasu retinowego RARE. Dzienne zapotrzebowanie ludzkiego organizmu na witaminę A jest określane na około 1 mg.

- W krajach, gdzie głównym składnikiem diety jest ryż, notuje się częste niedobory witaminy A. W celu umożliwienia zwiększenia spożycia witaminy A, metodami inżynierii genetycznej wyprodukowany został tzw. złoty ryż, zawierający geny z kukurydzy kodujące prowitaminę A.
- ZATRUCIA WITAMINĄ A:

- APOKAROTENOIDY - jest to grupa organicznych związków chemicznych, występujących powszechnie w wielu organizmach żywych. Powstają w wyniku oksydacyjnego rozkładu karotenoidów, katalizowanego przez oksygenazy karotenoidowe. Przykładami apokarotenoidów są: retinal, retinol, kwas retinowy i kwas abscysynowy.

β-Apo-8′-karotenal - jest to organiczny związek chemiczny z grupy apokarotenoidów.

Naturalny lub syntetyczny, pomarańczowożółty barwnik spożywczy. Jak inne karotenoidy, jest źródłem witaminy A, choć w o połowę mniejszym stopniu, niż beta-karoten. W przyrodzie występuje w wielu roślinach (głównie szpinaku oraz cytrusach), a także w wątrobie. Do celów przemysłowych wykorzystuje się głównie jego syntetyczną odmianę. Znajduje on zastosowanie głównie w przemyśle spożywczym, do barwienia takich produktów jak: kremowe serki do smarowania pieczywa, sery w plastrach, sery topione czy sosy sałatkowe. Jego dopuszczalne dzienne spożycie wynosi 2,5 mg/kg ciała. Jest uznawany za nieszkodliwy w zastosowaniach spożywczych, jedynie wysokie stężenie tego związku w organizmie może powodować żółte przebarwienia skóry.
β-Apo-8′-karotenian etylu - jest to organiczny związek chemiczny z grupy apokarotenoidów (podgrupa karotenoidów), ester etylowy kwasu beta-apo-8'-karotenowego.
Naturalny lub syntetyczny, pomarańczowożółty barwnik spożywczy. Występuje w małych ilościach w niektórych gatunkach roślin, ale do celów przemysłowyc wykorzystuje się często odmianę syntetyczną. Dopuszczalna dziennie spożycie wynosi 5 mg/kg masy ciała. ZASTOSOWANIE: Głównie w przemyśle spożywczym, do barwienia takich produktów jak przetworzona żywność lub mleczne napoje fermentowane, aromatyzowane i/lub z dodatkami smakowymi.
 Biksyna - jest to organiczny związek chemiczny z grupy apokarotenoidów. Naturalny, pomarańzowoczerwony barwnik terpenowy. Jest to ekstrakt otrzymywany z nasion arnoty właściwej (Bixa orellana) - gatunku niewielkiego drzewa z rodziny arnotowatych (Bixacea), który rośnie od północnej części Ameryki Południowej przez Amerykę Środkową po północny Meksyk - jest ona nazywana często annato. 5% tych nasion stanowią pigmenty, które zawierają 70-80% biksyny. Związek ten występuje w postaci dwóch izomerów geometrycznych, tj. jako cis- i trans-biksyna.
Biksyna - jest to organiczny związek chemiczny z grupy apokarotenoidów. Naturalny, pomarańzowoczerwony barwnik terpenowy. Jest to ekstrakt otrzymywany z nasion arnoty właściwej (Bixa orellana) - gatunku niewielkiego drzewa z rodziny arnotowatych (Bixacea), który rośnie od północnej części Ameryki Południowej przez Amerykę Środkową po północny Meksyk - jest ona nazywana często annato. 5% tych nasion stanowią pigmenty, które zawierają 70-80% biksyny. Związek ten występuje w postaci dwóch izomerów geometrycznych, tj. jako cis- i trans-biksyna. - Irony - jest to grupa organicznych związków chemicznych. Są to terpeny, a dokładniej 2-metylojonony, podobnie jak jonony, nie spełniają reguły izoprenowej. Jonony to organiczne związki chemiczne z grupy cyklicznych ketonów nienasyconych, zaliczane do terpenoidów. Nie są one jednak typowymi terpenoidami, ponieważ nie spełniają reguły izoprenowej, tzn. reszty izoprenowe nie są połączone ze sobą w sposób ogon - głowa. Irony występują w korzeniach irysów i w kwiatach akacji. Mają zapach fiołków. Używane są do wyrobu perfum. Najcenniejszą nutę daje (-)-trans-alfa-iron, natomiast nasilniejszym zapachem charakteryzuje się (+)-beta-iron, który jest też najbardziej rozpowszechniony w przyrodzie.
- Norbiksyna - jest to organiczny związek chemiczny z grupy karotenoidów, naturalny, barwnik, otrzymywany przez ekstrakcję nasion arnoty właściwej (Bixa orellana), nazywanej również annato.

- Perydynina - jest to organiczny związek chemiczny, karotenoid absorbujący światło, barwnik powiązany z chlorofilem w białku perydyninowo-chlorofilowym (PerCP), odpowiedzialny za absorpcję światła u bruzdnic (Dinoflagellata).
- KAROTENY- karoteny to grupa organicznych związków chemicznych, rozbudowanych przestrzennie węglowodorów nienasyconych zawierających 40 atomów węgla o wzorze sumarycznym C40H56. W cząsteczkach karotenów występują wieloatomowe układy sprzężonych wiązań podwójnych. W przyrodzie istnieją 3 izomery tego związku, różniące się umiejscowieniem jednego z wiązań podwójnych między atomami węgla. Karoten jest prowitaminą dla witaminy A (2 części karotenu to równowartość 1 części witaminy A). Występuje głównie jako alfa-karoten i beta-karoten. Istnieją także: gamma- delta- i epsilon- karoten. W 1928 roku Szwajcar Paul Karrer stwierdził, że beta-karoten jest głównym prekursorem witaminy A. Był to pierwszy przypadek ustalenia struktury prowitaminy. Następnie w 1930 roku ustalił wzór alfa-karotenu i zsyntetyzował go. W zależności od liczby i rodzaju pierścieni jonowych wyróżniamy alfa-, beta- i gamma-karoteny. Najbardziej znanym jest beta-karoten, żółty barwnik roślinny. Zawiera on dwa pierścienie beta-jononu, połączone tetramerem izoprenu. Alfa - Karoten zawiera po jednym pierścieniu alfa i beta-jononu, ma barwę czerwonobrązową. Gamma-karoten ma tylko jeden pierścień beta-jononu i występuje w grzybach i bakteriach, posiada barwę fioletową. Karoteny w żywieniu: Karoteny są pomarańczowożółtymi organicznymi barwnikami roślinnymi, występującymi m.in. w wielu warzywach i owocach. Zawartość karotenów w arzywach mieści się w granicach 0,5-31 mg/100g. Ważnymi źródłami karotenów dla człowieka są: bataty, brokuły 7-20 mg beta-karotenu/kg świeżej masy, buraki ćwikłowe, dynia 36 mg beta-karotenu/kg świeżej masy, kapusta włoska, kwiat nagietka, marchew 88-120 mg beta-karotenu/kg świeżej masy, morele, melony, pomidory 5 mg beta-karotenu/kg świeżej masy, papryka słodka czerwona 22 mg beta-karotenu/kg świeżej masy, sałata, ziele pietruszki. Dla glonożernych zwierząt akwariowych dobrym źródłem beta-karotenu jest spirulina. Częste spożywanie dużej ilości karotenów powoduje żółtawe zabarwienie skóry. Beta-karoten jako lek: Beta-Karoten jest bezpieczną odmianą karotenu, gdyż organizm przetwarza tylko taką jego ilość, jaka jest mu potrzebna. Przyczynia się między innymi do ochrony przed drobnoustrojami. Ma działanie antyoksydacyjne, a także korzystnie wpływa na funkcjonowanie wzroku, układu odpornościowego. W preparatach często podawany jest razem z witaminami E, D, B i wapniem.
 BETA KAROTEN - jest to organiczny związek chemiczny, prowitamina witaminy A i najbardziej aktywny jej izomer. Neutralizuje rodniki w organizmie, stosowany jest jako lek w chorobach wzroku i skóry. Właściwości farmakologiczne: Beta-karoten jest bezpieczną odmianą karotenu, gdyż organizm przetwarza tylko taką jego ilość, jaka jest mu potrzebna. Przyczynia się między innymi do ochrony przed drobnoustrojami. Ma działanie antyoksydacyjne, a także korzystnie wpływa na funkcjonowanie wzroku, systemu immunologicznego. W preparatach często podawany jest razem z witaminami E, D, B i wapniem. Beta-karoten był dość długo uważany za środek zapobiegający powstawaniu raka płuc. Późniejsze badania wykazały jednak, że u osób palących i narażonych na działanie azbestu spożywanie beta-karotenu minimalnie zwiększa, a nie zmniejsza ryzyko choroby nowotworowej. Prawdopodobnie jest to spowodowane tym, że beta-karoten w płucach wystawionych na działanie dymu papierosowego tworzy utlenione metabolity sprzyjające uszkodzeniom DNA. Spożywanie warzyw i owoców bogatych w karotenoidy zmniejsza jednak ryzyko raka, w tym raka płuc. Badania na fretkach wskazują, że beta-karoten w dużych i małych dawkach chroni przed metaplazją (metaplazja jest sytuacją, w której komórki zmieniły swój pierwotny, dojrzały typ zróżnicowania w inny typ zróżnicowanych komórek, również dojrzałych, w odpowiedzi adaptacyjnej na chroniczne podrażnienie, patogen lub karcynogen) w płucach, jeśli towarzyszą mu działające stabilizująco alfa-tokoferol i kwas askorbinowy. Długotrwała suplementacja diety w beta-karoten może w umiarkowany sposób ograniczać rozwój zmian umysłowych związanych z wiekiem. Beta-karoten nawet nie przekształcony w witaminę A chroni przed niekorzystnym wpływem wolnych rodników, które mogą wiązać się z substancjami znajdującymi się w komórkach i wywierać szkodliwy wpływ na błony komórkowe oraz na podział komórek. Redukuje również ujemne skutki radioterapii i chemioterapii. Zaobserwowano także, że może on spowodować powrót komórek dysplastycznych do normalnej funkcji. Wzmacnia system immunologiczny, chroni wyściółkę przewodu pokarmowego i dróg oddechowych przed infekcjami, zapobiega rozedmie płuc i bronchitowi (zapaleniu oskrzeli). Odgrywa istotną rolę w profilaktyce przeciwmiażdżycowej wpływając na obniżenie poziomu cholesterolu. Beta-karoten zapewnia ponadto prawidłowe funkcjonowanie narządu wzroku, zwłaszcza o zmierzchu, oraz warunkuje prawidłowe rogowacenie nabłonków, opóźnia procesy starzenia się. Wskazania: choroby skóry [fotodermatozy - choroby skóry spowodowane ekspozycją na światło słoneczne, związane z nadwrażliwością na promieniowanie UV, osutki (reakcje uczuleniowe) i pokrzywki świetlne, a także bielactwo], przygotowanie skóry na działanie promieniowania; zaburzenia widzenia o zmierzchu. Przeciwskazaniami są nadwrażliwość na lek i ciężka niewydolność wątroby i nerek. Działania niepożądne to m.in. bóle brzucha, biegunka, przejściowe żółte zabarwienie skóry.
BETA KAROTEN - jest to organiczny związek chemiczny, prowitamina witaminy A i najbardziej aktywny jej izomer. Neutralizuje rodniki w organizmie, stosowany jest jako lek w chorobach wzroku i skóry. Właściwości farmakologiczne: Beta-karoten jest bezpieczną odmianą karotenu, gdyż organizm przetwarza tylko taką jego ilość, jaka jest mu potrzebna. Przyczynia się między innymi do ochrony przed drobnoustrojami. Ma działanie antyoksydacyjne, a także korzystnie wpływa na funkcjonowanie wzroku, systemu immunologicznego. W preparatach często podawany jest razem z witaminami E, D, B i wapniem. Beta-karoten był dość długo uważany za środek zapobiegający powstawaniu raka płuc. Późniejsze badania wykazały jednak, że u osób palących i narażonych na działanie azbestu spożywanie beta-karotenu minimalnie zwiększa, a nie zmniejsza ryzyko choroby nowotworowej. Prawdopodobnie jest to spowodowane tym, że beta-karoten w płucach wystawionych na działanie dymu papierosowego tworzy utlenione metabolity sprzyjające uszkodzeniom DNA. Spożywanie warzyw i owoców bogatych w karotenoidy zmniejsza jednak ryzyko raka, w tym raka płuc. Badania na fretkach wskazują, że beta-karoten w dużych i małych dawkach chroni przed metaplazją (metaplazja jest sytuacją, w której komórki zmieniły swój pierwotny, dojrzały typ zróżnicowania w inny typ zróżnicowanych komórek, również dojrzałych, w odpowiedzi adaptacyjnej na chroniczne podrażnienie, patogen lub karcynogen) w płucach, jeśli towarzyszą mu działające stabilizująco alfa-tokoferol i kwas askorbinowy. Długotrwała suplementacja diety w beta-karoten może w umiarkowany sposób ograniczać rozwój zmian umysłowych związanych z wiekiem. Beta-karoten nawet nie przekształcony w witaminę A chroni przed niekorzystnym wpływem wolnych rodników, które mogą wiązać się z substancjami znajdującymi się w komórkach i wywierać szkodliwy wpływ na błony komórkowe oraz na podział komórek. Redukuje również ujemne skutki radioterapii i chemioterapii. Zaobserwowano także, że może on spowodować powrót komórek dysplastycznych do normalnej funkcji. Wzmacnia system immunologiczny, chroni wyściółkę przewodu pokarmowego i dróg oddechowych przed infekcjami, zapobiega rozedmie płuc i bronchitowi (zapaleniu oskrzeli). Odgrywa istotną rolę w profilaktyce przeciwmiażdżycowej wpływając na obniżenie poziomu cholesterolu. Beta-karoten zapewnia ponadto prawidłowe funkcjonowanie narządu wzroku, zwłaszcza o zmierzchu, oraz warunkuje prawidłowe rogowacenie nabłonków, opóźnia procesy starzenia się. Wskazania: choroby skóry [fotodermatozy - choroby skóry spowodowane ekspozycją na światło słoneczne, związane z nadwrażliwością na promieniowanie UV, osutki (reakcje uczuleniowe) i pokrzywki świetlne, a także bielactwo], przygotowanie skóry na działanie promieniowania; zaburzenia widzenia o zmierzchu. Przeciwskazaniami są nadwrażliwość na lek i ciężka niewydolność wątroby i nerek. Działania niepożądne to m.in. bóle brzucha, biegunka, przejściowe żółte zabarwienie skóry.
 LIKOPEN - jest to organiczny związek chemiczny z grupy karotenów, węglowodó® nienasycone o budowie podobnej do kauczuku naturalnego. Należy do rodziny naturalnych pigmentów (karotenoidów) występujących u roślin i zwierząt. Jest jednym z przeciwutleniaczy, posiada właściwości chroniące organizm przed licznymi chorobami układu krwionośnego, a przede wszystkim przed rakiem. Jest głównym karotenoidem, który (w odróżnieniu od beta-karotenu) po wchłonięciu w jelicie nie ulega przekształceniu do retinolu (nie jest substratem do cynkozależnej dioksygenazy, jak inne karotenoidy). WYSTĘPOWANIE: Likopen występuje obficie w pomidorach, przepękli indochińskiej (owoc stosowany jest w medycynie chińskiej, ale pojawiają się też doniesienia naukowe o jego działaniu przeciwnowotworowym. Owoce zawierają więcej likopenu 200-230mg likopenu/100g owoców niż pomidory, które zawierają 4,2mg likopenu/100g, a ponadto mają 10-krotnie więcej karotenu niż marchew. Ponadto owe substancje występują w łatwo przyswajalnej formie, z długołańcuchowymi kwasami tłuszczowymi), gujawie (tropikalne rośliny występujące na Karaibach, w Ameryce Środkowej i w północnej części Ameryki Południowej), melonach oraz innych czerwonych owocach (arbuzy, czerwone grapefruity, owoce dzikiej róży). Ze względu na łatwą rozpuszczalność w tłuszczach, najlepiej przyswajalny przez człowieka w postaci przetworzonej poprzez podgrzanie z oliwą. Na skalę przemysłową likopen jest ekstrahowany z pomidorów i czerwonych grejpfrutów. Likopen skutecznie oddziałuje na procesu nowotworzenia, jednak mechanizm tego zjawiska nie został do końca poznany. Sądzi się, że powoduje on odbudowanie połączeń międzykomórkowych, podobnie jak ma to miejsce w przypadku innych karotenoidów. Dowiedziono, że wysokie spożycie pomidorów wywołuje efekt ochronny przeciwko nowotworom przewodu pokarmowego oraz redukuje o 50% ryzyko zgonu na podłożu nowotworowym u ludzi w wieku podeszłym. Pomidory i inne produkty zawierające likopen należy szczególnie polecać mężczyznom będącym w grupie ryzyka wystąpienia raka prostaty lub już walczącym z tą chorobą. Jak pokazują badania spożycie 10 porcji pomidorów w tygodniu może prowadzić do obniżenia ryzyka wystąpienia raka prostaty o aż 35%. Natomiast u kobiet spożycie produktów bogatych w likopen zmniejsza ryzyko wystąpienia raka piersi. Należy zaznaczyć, iż dotychczas żadne badania nie wskazywały na negatywny efekt spożycia nawet dużych ilości pomidorów lub innych produktów bogatych w likopen. Związek ten posiada również zdolność przeciwdziałania agregacji płytek krwi i tworzeniu skrzepów oraz zapobiega rozwojowi miażdżycy i chorób sercowo-naczyniowych. Ponadto wykazano, że spożywanie produktów bogatych w likopen może obniżać ryzyko ostrego zawału mięśnia sercowego, wystąpienia choroby wieńcowej, jak również może zmniejszyć liczbę zgonów z powodu choroby wieńcowej. Likopen znajduje zastosowanie jako barwnik w przemyśle spożywczym. Barwi się nim np. batoniki odżywcze, zupy, jogurty, napoje gazowane oraz produkty żelujące.
LIKOPEN - jest to organiczny związek chemiczny z grupy karotenów, węglowodó® nienasycone o budowie podobnej do kauczuku naturalnego. Należy do rodziny naturalnych pigmentów (karotenoidów) występujących u roślin i zwierząt. Jest jednym z przeciwutleniaczy, posiada właściwości chroniące organizm przed licznymi chorobami układu krwionośnego, a przede wszystkim przed rakiem. Jest głównym karotenoidem, który (w odróżnieniu od beta-karotenu) po wchłonięciu w jelicie nie ulega przekształceniu do retinolu (nie jest substratem do cynkozależnej dioksygenazy, jak inne karotenoidy). WYSTĘPOWANIE: Likopen występuje obficie w pomidorach, przepękli indochińskiej (owoc stosowany jest w medycynie chińskiej, ale pojawiają się też doniesienia naukowe o jego działaniu przeciwnowotworowym. Owoce zawierają więcej likopenu 200-230mg likopenu/100g owoców niż pomidory, które zawierają 4,2mg likopenu/100g, a ponadto mają 10-krotnie więcej karotenu niż marchew. Ponadto owe substancje występują w łatwo przyswajalnej formie, z długołańcuchowymi kwasami tłuszczowymi), gujawie (tropikalne rośliny występujące na Karaibach, w Ameryce Środkowej i w północnej części Ameryki Południowej), melonach oraz innych czerwonych owocach (arbuzy, czerwone grapefruity, owoce dzikiej róży). Ze względu na łatwą rozpuszczalność w tłuszczach, najlepiej przyswajalny przez człowieka w postaci przetworzonej poprzez podgrzanie z oliwą. Na skalę przemysłową likopen jest ekstrahowany z pomidorów i czerwonych grejpfrutów. Likopen skutecznie oddziałuje na procesu nowotworzenia, jednak mechanizm tego zjawiska nie został do końca poznany. Sądzi się, że powoduje on odbudowanie połączeń międzykomórkowych, podobnie jak ma to miejsce w przypadku innych karotenoidów. Dowiedziono, że wysokie spożycie pomidorów wywołuje efekt ochronny przeciwko nowotworom przewodu pokarmowego oraz redukuje o 50% ryzyko zgonu na podłożu nowotworowym u ludzi w wieku podeszłym. Pomidory i inne produkty zawierające likopen należy szczególnie polecać mężczyznom będącym w grupie ryzyka wystąpienia raka prostaty lub już walczącym z tą chorobą. Jak pokazują badania spożycie 10 porcji pomidorów w tygodniu może prowadzić do obniżenia ryzyka wystąpienia raka prostaty o aż 35%. Natomiast u kobiet spożycie produktów bogatych w likopen zmniejsza ryzyko wystąpienia raka piersi. Należy zaznaczyć, iż dotychczas żadne badania nie wskazywały na negatywny efekt spożycia nawet dużych ilości pomidorów lub innych produktów bogatych w likopen. Związek ten posiada również zdolność przeciwdziałania agregacji płytek krwi i tworzeniu skrzepów oraz zapobiega rozwojowi miażdżycy i chorób sercowo-naczyniowych. Ponadto wykazano, że spożywanie produktów bogatych w likopen może obniżać ryzyko ostrego zawału mięśnia sercowego, wystąpienia choroby wieńcowej, jak również może zmniejszyć liczbę zgonów z powodu choroby wieńcowej. Likopen znajduje zastosowanie jako barwnik w przemyśle spożywczym. Barwi się nim np. batoniki odżywcze, zupy, jogurty, napoje gazowane oraz produkty żelujące.
KSANTOFILE - jest to grupa organicznych związków chemicznych, barwników roślinnych należących do karotenoidów, tlenowych pochodnych karotenów. Ksantofile są pomocniczymi barwnikami fotosyntezy. Pełnią także funkcję przeciwutleniaczy, przez co chronią komórkę, a zwłaszcza chloroplasty przed szkodliwym działaniem reaktywnych form tlenu. Dzięki wielu sprzężonym wiązaniom podwójnym nadają żółtą, pomarańczową lub czerwoną barwę kwiatom, owocom, a taże żółtku jaj (luteina). Do częściej występujących należą fukoksantyna i wiolaksantyna oraz zeaksantyna (odpowiedzialna za barwę kukurydzy). BIOSYNTEZA: Ksantofile powstają drogą bezpośredniego wprowadzenia tlenu do cząsteczki z udziałem oksygenaz mieszanych. Najczęściej spotykane w naturze są pochodne hydroksylowe (grupa -OH, najczęściej w pozycjach C-3 i C-3' cząsteczki), chociaż znane są też ksantofile zawierające w cząsteczce grupy aldehydowe, ketonowe, karboksylowe i hydroksymetylowe (-CH2OH). Ze względu na różnorodność pochodnych tlenowych należy sądzić, że hydroksylacja zachodzi zarówno przed, jak i po cyklizacji.
Źródło: Wikipedia. Żółtawe lub brunatne ksantofile
odpowiedzialne są za jesienne żółte zabarwienie liści.
 *Anteraksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Naturalny żółty barwnik u glonów, mogący pełnić funkcję barwnika pomocniczego w kompleksach zbierających światło. Energia wzbudzenia z karotenoidów przenoszona jest na chlorofil w centrum reakcji fotoukładu. U roślin wyższych i większości glonów anteraksantyna jest związkiem uczestniczącym w cyklu ksantofilowym, pośrednim produktem przemiany wiolaksantyny w zeaksantynę. Cykl ksantofilowy umożliwia rozproszenie nadmiaru energii, chroniąc aparat fotosyntetyczny przed uszkodzeniem.
*Anteraksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Naturalny żółty barwnik u glonów, mogący pełnić funkcję barwnika pomocniczego w kompleksach zbierających światło. Energia wzbudzenia z karotenoidów przenoszona jest na chlorofil w centrum reakcji fotoukładu. U roślin wyższych i większości glonów anteraksantyna jest związkiem uczestniczącym w cyklu ksantofilowym, pośrednim produktem przemiany wiolaksantyny w zeaksantynę. Cykl ksantofilowy umożliwia rozproszenie nadmiaru energii, chroniąc aparat fotosyntetyczny przed uszkodzeniem.
 *Astaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest metabolitem zeaksantyny i kataksantyny. Jak wiele innych karotenoidów, rozpuszcza się w tłuszczach. Cząsteczka astaksantyny zawiera liczne sprzężone wiązania wielokrotne, warunkujące barwę substancji i jej właściwości przeciwutleniające. Z powodu występowania dwóch centrów chiralności (grupy hydroksylowe w pozycjach 3 i 3' pierścieni), ma trzy diastereoizomery: 3R,3'R, 3R,3'S (mezo) i 3S,3'S. Występowanie: Występuje w ciele jednokomórkowych glonów, drożdży, łososi, pstrągów, kryli, krewetek, raków i innych skorupiaków, a także w piórach niektórych ptaków. Jest odpowiedzialna za czerwony kolor mięsa łososia i krewetek. Powszechnie występuje w ciele kriofilnych glonów, takich jak zawłotnia śnieżna. Pełni u nich funkcję filtra chroniącego przed ultrafioletem. Występując w dużych zagęszczeniach na powierzchni nadtopionego śniegu, glony te powodują jego zakwit, znany jako czerwony śnieg. ZASTOSOWANIE: Astaksantyna, w przeciwieństwie do wielu innych karotenoidów, nie ulega w organizmie człowieka przekształceniu do witaminy A. Podobnie jak inne karotenoidy jest w ograniczonym stopniu przyswajana drogą pokarmową. W badaniach klinicznych nie stwierdzono szkodliwych efektów spożywania astaksantyny. Jest naturalnym składnikiem odżywczym, stosuje się ją także jako dodatek do żywności. Uzyskiwana jest zarówno syntetycznie, jak i ze źródeł naturalnych. Otrzymywanie: źródła naturalne: na skalę przemysłową astaksantynę uzyskuje się z następujących organizmów: kryl pacyficzny (Euphausia pacifica), kryl antarktyczny (Euphausia superba), Haematococcus pluvialis (zielenica), krewetka północna (Pandalus borealis). Głównym naturalnym źródłem astaksantyny są mikroalgi Haematococcus pluvialis, w których zawartość tego związku jest najwyższa wśród znanych organizmów. Z kilograma suchej masy tych glonów otrzymać można ponad 40 gramów astaksantyny. Udział poszczególnych diastereoizomerów w substancji naturalnej jest różny w zależności od źródła. Na przykład produkowana przez Haematococcus pluvialis, zawiera głównie izomer (3S,3'R)-astaksantyna.
*Astaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest metabolitem zeaksantyny i kataksantyny. Jak wiele innych karotenoidów, rozpuszcza się w tłuszczach. Cząsteczka astaksantyny zawiera liczne sprzężone wiązania wielokrotne, warunkujące barwę substancji i jej właściwości przeciwutleniające. Z powodu występowania dwóch centrów chiralności (grupy hydroksylowe w pozycjach 3 i 3' pierścieni), ma trzy diastereoizomery: 3R,3'R, 3R,3'S (mezo) i 3S,3'S. Występowanie: Występuje w ciele jednokomórkowych glonów, drożdży, łososi, pstrągów, kryli, krewetek, raków i innych skorupiaków, a także w piórach niektórych ptaków. Jest odpowiedzialna za czerwony kolor mięsa łososia i krewetek. Powszechnie występuje w ciele kriofilnych glonów, takich jak zawłotnia śnieżna. Pełni u nich funkcję filtra chroniącego przed ultrafioletem. Występując w dużych zagęszczeniach na powierzchni nadtopionego śniegu, glony te powodują jego zakwit, znany jako czerwony śnieg. ZASTOSOWANIE: Astaksantyna, w przeciwieństwie do wielu innych karotenoidów, nie ulega w organizmie człowieka przekształceniu do witaminy A. Podobnie jak inne karotenoidy jest w ograniczonym stopniu przyswajana drogą pokarmową. W badaniach klinicznych nie stwierdzono szkodliwych efektów spożywania astaksantyny. Jest naturalnym składnikiem odżywczym, stosuje się ją także jako dodatek do żywności. Uzyskiwana jest zarówno syntetycznie, jak i ze źródeł naturalnych. Otrzymywanie: źródła naturalne: na skalę przemysłową astaksantynę uzyskuje się z następujących organizmów: kryl pacyficzny (Euphausia pacifica), kryl antarktyczny (Euphausia superba), Haematococcus pluvialis (zielenica), krewetka północna (Pandalus borealis). Głównym naturalnym źródłem astaksantyny są mikroalgi Haematococcus pluvialis, w których zawartość tego związku jest najwyższa wśród znanych organizmów. Z kilograma suchej masy tych glonów otrzymać można ponad 40 gramów astaksantyny. Udział poszczególnych diastereoizomerów w substancji naturalnej jest różny w zależności od źródła. Na przykład produkowana przez Haematococcus pluvialis, zawiera głównie izomer (3S,3'R)-astaksantyna. 
ŹRÓDŁA SZTUCZNE: Większość astaksantyny produkowana jest za pomocą syntezy chemicznej. Wartość tej produkcji wynosi około 200 mln dolarów, a cena sprzedaży to około 5-6 tysięcy dolarów za kilogram (lipiec 2012). W typowym podejściu astaksantynę otrzymuje się w wieloetapowej syntezie, której końcowym etapem jest reakcja Wittiga pomiędzy ylidem zawierającym końcowe pierścienie izoforonowe a symetrycznym nienasyconym dialdehydem budującym centralny łańcuch cząsteczki.

W zależności od użytych substratów, produkt końcowy może być mieszaniną 3 diastereoizomerów (3R, 3' R, 3R,3'S (mezo) i 3S, 3'S w proporcji statystycznej 1:2:1) lub być związkiem stereochemicznie czystym o wybranej konfiguracji. Produkt syntetyczny dostępny handlowo jest mieszaniną izomerów.
*Cytraksantyna - jest to organiczny związek chemiczny z grupy ksantofili, naturalny barwnik spożywczy otrzymywany z różnych gatunków suszonych roślin. Jego dopuszczalne dzienne spożycie wynosi 0,4 mg/kg masy ciała.
*Flawoksantyna - jest to organiczny związek chemiczny z grupy ksantofili (podgrupa karotenoidów). Naturalny, żółtozłoty barwnik spożywczy. Jego niewielkie ilości można znaleźć w wielu gatunkach roślin. Dopuszczalne dzienne spożycie wynosi 5 mg/kg masy ciała. Do celów przemysłowych uzyskuje się ją z jaskrów. Dziś używana jest rzadko; wyłącznie w przemyśle cukierniczym.
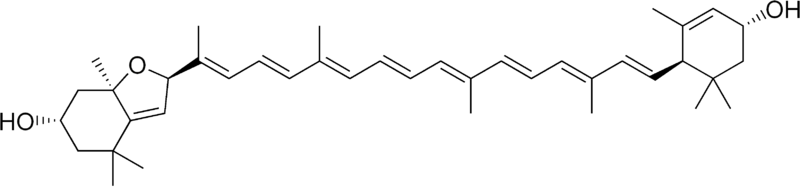
*Fukoksantyna (łac. fucus - morszczyn, gr. ksanthos - żółty) - jest to organiczny związek chemiczny z grupy ksantofili, naturalny ciemnoczerwony lub brązowy barwnik, występujący w komórkach okrzemek oraz brunatnic. W połączeniu z chlorofilem (jeśli jest go dostatecznie dużo) nadaje komórkom brunatne zabarwienie. Fukoksantyna absorbuje światło od żółto-zielonej do żółto-niebieskiej części spektrum.
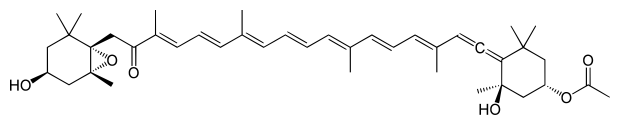
*Kantaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Naturalny, różowy barwnik spożywczy. Obecny jest w małych ilościach w wielu gatunkach roślin oraz grzybów, a także w piórach niektórych ptaków. Można go wyprodukować syntetycznie z Beta-karotenu, ale na skalę przemysłową jest otrzymywany z piór flamingów. Jego użycie do celów spożywczych jest zakazane w niektórych krajach. Zastosowanie: W przemyśle spożywczym używana do barwienia paluszków rybnych, lodów, ptasiego mleczka, a także marynat, sosów i konserw. Znajduje również zastosowanie do produkcji tabletek opalających. Uwaga: Przy stosowaniu tabletek opalających zawierających kantaksantynę mogą wystąpić problemy ze wzrokiem (np. utrata nocnego widzenia), a także: odbarwienia skóry, nawracające pokrzywki oraz wrażliwość na ostre światło. W skrajnych przypadkach kantaksantyna może być przyczyną retinopatii barwnikowej.

 *Kryptoksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest naturalnym pigmentem, występującym w przyrodzie. Został wyizolowany z płatków i owoców miechunki (Physalis L. - rodzaju roślin z rodziny psiankowatych, którego gatunki pochodzą z Ameryki Południowej). Rośliny też używa się również do produkcji pigmentu do celów przemysłowych. Po krystalizacji z mieszaniny benzenu i metanolu przyjmuje postać ciemnoczerwonych słupków, natomiast z mieszaniny metanolu i etery dietylowego wykrystalizowuje tworząc czerwone płatki o metalicznym połysku. Dopuszczalne dzienne spożycie wynosi 5 mg/kg.
*Kryptoksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest naturalnym pigmentem, występującym w przyrodzie. Został wyizolowany z płatków i owoców miechunki (Physalis L. - rodzaju roślin z rodziny psiankowatych, którego gatunki pochodzą z Ameryki Południowej). Rośliny też używa się również do produkcji pigmentu do celów przemysłowych. Po krystalizacji z mieszaniny benzenu i metanolu przyjmuje postać ciemnoczerwonych słupków, natomiast z mieszaniny metanolu i etery dietylowego wykrystalizowuje tworząc czerwone płatki o metalicznym połysku. Dopuszczalne dzienne spożycie wynosi 5 mg/kg.
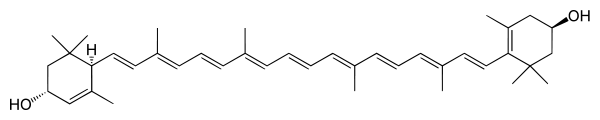 *Luteina - związek organiczny, żółty barwnik należący do ksantofili, alkoholowa pochodna alfa-karotenu. Do zastosowań przemysłowych otrzymuje się ją z traw. W stanie naturalnym występuje m.in. w żółtku i w komórkach tłuszczowych. Ponadto luteinę zawiera kwiat aksamitek, kwiat słonecznika, ziele lucerny, ziele pokrzywy. Luteina jest istotna dla prawidłowego funkcjonowania narządu wzroku, gdyż chroni go przed uszkodzeniami powodowanymi przez wolne rodniki. W przemyśle spożywczym używa się jej jako barwnika do zup i napojów alkoholowych, a także ciast i przetworzonej żywności. Luteina jest także składnikiem niektórych karm dla zwierząt (np. drobiu).
*Luteina - związek organiczny, żółty barwnik należący do ksantofili, alkoholowa pochodna alfa-karotenu. Do zastosowań przemysłowych otrzymuje się ją z traw. W stanie naturalnym występuje m.in. w żółtku i w komórkach tłuszczowych. Ponadto luteinę zawiera kwiat aksamitek, kwiat słonecznika, ziele lucerny, ziele pokrzywy. Luteina jest istotna dla prawidłowego funkcjonowania narządu wzroku, gdyż chroni go przed uszkodzeniami powodowanymi przez wolne rodniki. W przemyśle spożywczym używa się jej jako barwnika do zup i napojów alkoholowych, a także ciast i przetworzonej żywności. Luteina jest także składnikiem niektórych karm dla zwierząt (np. drobiu).
 *Rodoksantyna -
*Rodoksantyna -
jest to organiczny związek chemiczny z grupy ksantofili. Naturalny barwnik spożywczy. Jego niewielkie ilości można znaleźć w wielu gatunkach roślin, np. cisu (Taxus baccata) i piórach niektórych ptaków (np. z rodzaju Ptillinopus i rodziny bławatników). Dopuszczalne dzienne spożycie wynosi 5 mg/kg masy ciała.
 *Rubiksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest to naturalny, żółtopomarańczowy lub pomarańczowoczerwony barwnik spożywczy. Do celów produkcyjnych uzyskuje się go z płatków roślin z rodziny różowatych. Chociaż początkowo sądzono, że barwnik jest specyficzny dla rodzaju Rosa to kolejna badania wykazały jego obecność także u innych roślin. Poza płatkami kwiatów, rubksantyna może występować także w owocach, co potwierdzono u goździkowca jednokwiatowego, którego owoce są spożywane w Ameryce Łacińskiej. Barwnik wykryto również u bakterii Staphylococcus aureus. Dopuszczalne dzienne spożycie wynosi 5 m/kg masy ciała.
*Rubiksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest to naturalny, żółtopomarańczowy lub pomarańczowoczerwony barwnik spożywczy. Do celów produkcyjnych uzyskuje się go z płatków roślin z rodziny różowatych. Chociaż początkowo sądzono, że barwnik jest specyficzny dla rodzaju Rosa to kolejna badania wykazały jego obecność także u innych roślin. Poza płatkami kwiatów, rubksantyna może występować także w owocach, co potwierdzono u goździkowca jednokwiatowego, którego owoce są spożywane w Ameryce Łacińskiej. Barwnik wykryto również u bakterii Staphylococcus aureus. Dopuszczalne dzienne spożycie wynosi 5 m/kg masy ciała.  *Wiolaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Naturalny, pomarańczowy barwnik spożywczy, występujący w kwiatach fiołków (Viola). Ksantofile są częścią aparatu fotosyntetycznego roślin wyższych, mogą być zarówno barwnikami pomocniczymi absorbującymi energię świetlną, jak i elemntami strukturalnymi kompleksów zbierających światło. Energia wzbudzenia może być przenoszona z wiolaksantyny z wydajnością około 54%. Także u glonów stwierdzono przyczynianie się wiolaksantyny do absorpcji światła. Wiolaksantyna bierze udział w cyklu ksantofilowym chroniącym rośliny przed nadmiarem światła. Wzajemne przekształcenie w zeaksantynę odbywa się przy udziale deepoksydazy wiolaksatyny i epoksydazy zeaksantyny. Produktem pośrednim jest anteraksantyna. Cykl ksantofilowy zapewnia ochronę przed nadmiarem światła roślinom oraz glonom. Przekształcenie wiolaksantyny w zeaksantynę wiąże się ze zmianami konformacyjnymi w kompleksach LHC (kompleksy zbierające energię świetlną) co umożliwia wygaszenie fluorescencji przez rozpraszanie ciepła. Wiolaksantyna jest też substratem dla szlaku biosyntezy kwasu abscysynowego. Wiolaksantyna jest także stosowana (choć już rzadko) do barwienia żywności. Dopuszczalne dzienne spożycie wynosi 5 mg/kg ciała.
*Wiolaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Naturalny, pomarańczowy barwnik spożywczy, występujący w kwiatach fiołków (Viola). Ksantofile są częścią aparatu fotosyntetycznego roślin wyższych, mogą być zarówno barwnikami pomocniczymi absorbującymi energię świetlną, jak i elemntami strukturalnymi kompleksów zbierających światło. Energia wzbudzenia może być przenoszona z wiolaksantyny z wydajnością około 54%. Także u glonów stwierdzono przyczynianie się wiolaksantyny do absorpcji światła. Wiolaksantyna bierze udział w cyklu ksantofilowym chroniącym rośliny przed nadmiarem światła. Wzajemne przekształcenie w zeaksantynę odbywa się przy udziale deepoksydazy wiolaksatyny i epoksydazy zeaksantyny. Produktem pośrednim jest anteraksantyna. Cykl ksantofilowy zapewnia ochronę przed nadmiarem światła roślinom oraz glonom. Przekształcenie wiolaksantyny w zeaksantynę wiąże się ze zmianami konformacyjnymi w kompleksach LHC (kompleksy zbierające energię świetlną) co umożliwia wygaszenie fluorescencji przez rozpraszanie ciepła. Wiolaksantyna jest też substratem dla szlaku biosyntezy kwasu abscysynowego. Wiolaksantyna jest także stosowana (choć już rzadko) do barwienia żywności. Dopuszczalne dzienne spożycie wynosi 5 mg/kg ciała.
 *Zeaksantyna - jest
*Zeaksantyna - jest to organiczny związek chemiczny z grupy ksantofili. Jest jednym z dwóch karotenoidów znajdujących się w siatkówce oka. Naturalnie występuje w brokułach, brukselce, kukurydzy i pomidorach. Zawiera ją także ziele fiołka trójbarwnego i polnego.
INNE BARWNIKI FOTOSYNTETYCZNE:





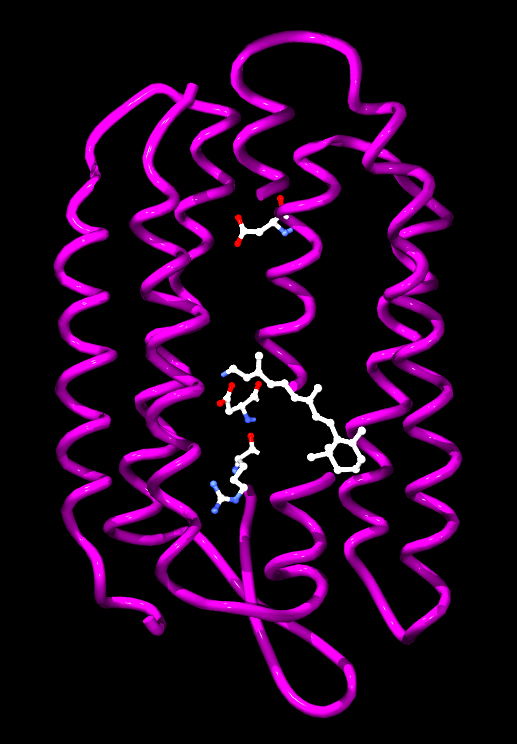 |
Źródło: Wikipedia. Model cząsteczki bakteriorodopsyny
z Halobacterium salinarum z oznaczonymi aminokwasami
biorącymi udział w przenoszeniu protonu oraz retinalem.
|
1) Bakteriorodopsyna - jest to białko o masie 26 kDa występujące u halobakterii (klasy archeonów w typie Euryarchaeota, które zostały znalezione w wodach o zasoleniu zbliżonym do roztworu nasyconego. Zalicza się je do organizmów określanych jako halofile. Występują w większości środowisk o dużym zasoleniu, wilgotności i dostępności materii organicznej) należących do domeny archeowców. Zalicza się do białek określanych jako pompy protonowe, posiada zdolność przenoszenia protonów przez błonę komórkową pod wpływem światła. Wytworzona różnica stężeń jonów wodorowych wykorzystywana jest następnie do syntezy ATP. Część naukowców proces syntezy ATP przy udziale bakteriorodopsyny traktuje jako rodzaj fotosyntezy. Sztucznie wytworzone struktury zawierające błony z bakteriorodopsyną oraz syntazą ATP pochodzącą z mitochondriów posłużyły do doświadczalnego potwierdzenia chemiosmotycznej teorii Mitchella. Bakteriorodopsyna jest integralnym białkiem błonowym zwykle tworzącym dwuwymiarowe plamy, określane jako "purpurowa błona", których powierzchnia może obejmować do 50% powierzchni komórki archeowców. Powtarzające się elementy sześciokątnej siatki składają się z trzech identycznych łańcuchów białkowych, każdego obróconego o 120 stopni w stosunku do poprzedniego. Każdy łańcuch ma siedem transmembranowych alfa helis i zawiera jedną cząsteczkę retinalu. Trzeciorzędowa struktura bakteriorodopsyny przypomina rodopsynę kręgowców, barwnik odpowiedzialny za reakcję na światło, obecny w siatkówce oka. Pomimo podobnej funkcji obu barwników, sekwencje aminokwasów łańcuchów białkowych znacznie się różnią. Zarówno rodopsyna, jak i bakteriorodopsyna należą do rodziny receptorów białkowych 7TM, jednak rodopsyna należy do receptorów sprzężonych z białkiem G (Receptory sprzężone z białkami G - GPCR - z ang G PROTEIN - COUPLED RECEPTOR - RECEPTORY SIEDMIOTRANSBŁONOWE, 7TM - to rodzaj transmembranowych receptorów metabotropowych, czyli regulujących funkcjonowanie kanałów jonowych, które reagują na sygnały docierające do komórki za pośrednictwem neuroprzekaźników aktywując białko G związane z receptorem po drugiej stronie błony komórkowej), bakteriorodopsyna zaś nie. Struktura bakteriorodopsyny została poznana dzięki rentgenografii strukturalnej i opisana po raz pierwszy w 1990 roku. Bakteriorodopsyna ma barwę fioletową, a maksimum absorpcji przypada na długości fal odpowiadające barwie zielonej (długość fali 500-650 nm, z maksimum absorpcji w 568 nm). Pochłonięcie fotonu przez cząsteczkę retinalu prowadzi do zmian konformacyjnych części białkowej, których efektem jest przeniesienie protonu przez błonę komórkową. Grupa chromoforowa przyłączona jest kowalencyjnie do Lys216, tworząc zasadę Schiffa. Po fotoizomeryzacji cząśteczki retinalu, proton przenoszony jest na Asp85. Miejsce przyłączenia protonu znajduje się po stronie zewnątrzkomórkowej. Retinal powraca do pierwotnej postaci w wyniku pobrania protonu z Asp96 znajdującej się po stronie wewnątrzkomórkowej, a Asp96 pobiera proton z wnętrza komórki.

Źródło: Wikipedia. Zmiany konformacyjne pod wpływem światła.
W efekcie stężenie protonów wzrasta w przestrzeni zewnątrzkomórkowej i maleje wewnątrz komórki. Znane są również inne białka o podobnych do bakteriorodopsyny właściwościach; przenosząca jony chlorkowe halorodopsyna oraz aktywowane przez światło kanały jonowe jak ChR1 i ChR2. Aparat fotosyntetyczny niemal wszystkich organizmów fotosyntetyzujących oparty jest na chlorofilu lub bakteriochlorofilu. Pomimo zastosowania innego barwnika mechanizm przekształcania energii świetlnej na energię wiązań chemicznych na halobakterii jest podobny. W pierwszej fazie wytwarzany jest gradient protonowy, a w drugiej energia gradientu wykorzystywana przez syntezę ATP. W przypadku chlorofilu możliwe jest wykorzystanie jednocześnie wielu barwników absorbujących światło obecnych w antenach fotosyntetycznych, proces oparty na bakteriorodopsynie umożliwia wykorzystanie jedynie długości fal absorbowanych przez białko. Jest to jedyna forma fotosyntezy obecna u archeowców. Nie jest jednak połączona z asymilacją dwutlenku węgla.
 |
| Źródło: Wikipedia. Fotoizomeryzacja cząsteczki retinalu. |
2) Halorodopsyna - jest inegralnym białkiem błonowym występującym u halobakterii należących do domeny archeowców. Ma zdolność przenoszenia przez błonę komórkową pod wpływem światła jonów chlorkowych lub protonów. Przy oświetlaniu światłem zielonym halorodopsyna przenosi przez błonę jony chlorkowe, a przy dodatkowym oświetleniu światłem niebieskim zamienia się w pompę protonową. Jej budowa zbliżona jest do bakteriorodopsyny. Sekwencja aminokwasów w tych białkach pokrywa się w 30%. Także struktura oraz mechanizm działania zbliżony jest do bakteriorodopsyny pełniącej jedynie funkcję pompy protonowej. Zmiana we właściwościach, polegająca na nabyciu zdolności do przenoszenia jonów chlorkowych, powstała prawdopodobnie w wyniku zamiany w bakteriorodopsynie asparaginianu w pozycji 85 na treoninę. Działanie halorodopsyny prowadzi do wytworzenia różnicy pH w poprzek błony. Energia gradientu elektrochemicznego wykorzystywana jest następnie do syntezy ATP.
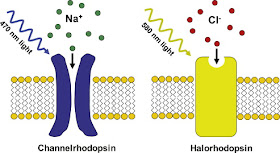 |
| Źródło: Bio-associate.blogspot.com
3) Fikobiliny (gr. fikos - glon, łac. bilis - żółć) - są to chromofory występujące u sinic oraz w chloroplastach glaukocystofitów, krasnorostów i kryptomonad. Fikobiliny, jako jedyne barwniki fotosyntetyczne są rozpuszczalne w wodzie, w połączeniu z odpowiednimi białkami tworzą struktury antenowe zwane fikobilisomami, które przekazują energię pochłoniętych fotonów na cząsteczki chlorofili fotoukładu II. Fikobiliny szczególnie wydajnie absorbują światło czerwone, pomarańczowe, żółte i zielone, czyli w zakresie długości fali częściowo nieabsorbowanym przez chlorofile. Organizmy żyjące w wodach płytkich posiadają zazwyczaj fikobiliny absorbujące światło żółte i czerwone, natomiast żyjące w wodach głębszych - światło zielone. Fikobiliny wykazują fluorescencję i są często wykorzystywane w technikach immunofluorescencyjnych jako znaczniki fluorescencyjne przyłączane do przeciwciał. Znane są cztery typy fikobilin: fikocyjanobilina (niebieska), fikoerytrobilina (czerwona), fikourobilina (pomarańczowa), fikowiolobilina. Fikobiliny te występują w różnych kombinacjach z fikobiliproteinami, tworząc np. fikocyjaninę lub fikoerytrynę. Budowa: Z chemicznego punktu widzenia fikobiliny zbudowane są na podstawie szkieletu tetrapirolowego - otwartego łańcucha czterech pierścieni pirolowych. Podobny szkielet tetrapirolowy ma bilirubina występująca w żółci (bilirubina, podobnie jak fikobiliny, jest światłoczuła, co wykorzystuje się w fototerapii żółtaczki noworodków) oraz fitochrom, natomiast w chlorofilu i hemie zbudowane są z czterech cząsteczek pirolu zamkniętych w pierścień porfirynowy.

Źródło: Wikipedia.
4) Fikocyjanina - niebieski barwnik, występujący u krasnorostów, kryptofitów i sinic. Ma właściwości fluorescencyjne i antyoksydacyjne. Barwnik jest kompleksem chromoforu nazywanego fikocyjanobiliną i białka. Fikocyjanina wchodzi w skład fikobilisomów.
5) Fikoerytryna (gr. phykos - trawa morska, wodorost, eryhros - czerwony) - jest to czerwony barwnik występujący u krasnorostów, kryptofitów i sinic. Barwnik jest kompleksem chromoforu nazywanego fikoerytrobiliną i białka. Fikoerytryna wchodzi w skład fikobilisomów.

Źródło: Wikipedia. Fikoerytrynobilina.
6) Proteorodopsyna - jest to białko występujące u morskich organizmów planktonowych, należących do bakterii, archeowców i eukariotów. Jest białkiem transbłonowym zawierającym retinal i pełni funkcję pompy protonowej. Właściwości białka mogą być wykorzystywane przez organizmy do wytwarzania ATP w formie fotoautotrofizmu zbliżonego do fotosyntezy. Znanych jest wiele wariantów proteorodopsyny o różnych widmach absorpcji. U części organizmów białko spełnia funkcje sensoryczne. Geny umożliwiające wykorzystanie proteorodopsyny rozpowszechniły się wśród organizmów planktonicznych w wyniku poziomego transferu genów. Prawdopodobnie przeniesienie genów proterodopsyny jest stosunkowo częstym i skoordynowanym wydarzeniem ewolucyjnym. Odkrycie: Proteorodopsyna została odkryta w roku 2000. Początkowo stwierdzono jej obecność w błonach komórkowych gamma-proteobakterii występujących we wschodniej i północnej części Oceanu Spokojnego, a następnie również u mikroorganizmów żyjących w północnej cześci Oceanu Atlantyckiego, w Oceanie Południowym i Arktycznym. Odmienne waruanty genu kodującego proteorodopsynę stwierdzono u organizmów żyjących w Morzu Śródziemnym, Morzu Czerwonym, Morzu Sargassowym i Morzu japońskim. Wykazano też obecność proteorodopsyny u bakterii powszechnie występującej w morzach. Warianty proteorodopsyny z różnych lokalizacji charakteryzują się odmiennym rozkładem maksimów absoprcji umożliwiając dostosowanie się organizmów do warunków oświetleniowych zależnych od położenia geograficznego i głębokości. Budowa i działanie: Proteorodopsyna wykazuje duże podobieństwo do bakteriorodopsyny, występującej u archeowców. Centrum aktywne bakteriorodopsyny Asp 82, Asp85 (pierwotny akceptor protonu), Asp212 i Lys216 (miejsce wiązania retinalu) jest zachowane w proteorodopsynie jako Arg94, Asp97 i Lys231. Brak jest jednak występującego w bakteriorodopsynie kwasu karboksylowego w postaci Glu194 lub Glu204, który prawdopodobnie jest odpowiedzialny za uwalnianie protonu po zewnętrznej stronie błony komórkowej. Proterodopsyna w komórkach organizmów planktonicznych spełnia funkcje pompy protonowej napędzanej światłem. Umożliwia przetrwanie organizmom żyjącym w wierzchniej warstwie oceanów, w warunkach niedostatku substancji pokarmowych. Doświadczalnie potwierdzono, że światło powoduje wzmożony rozwój bakterii w porównaniu z bakteriami hodowanymi w ciemności. Chromofor w postaci retinalu jest kowalencyjnie związane z apoproteiną (apoenzymem - białkową częścią enzymu, która po połączeniu z odpowiednimi koenzymami lub grupami prostetycznymi tworzy holoenzym) tworząc zasadę Schiffa z udziałem Lys231. W wyniku oświetlenia zmianie ulega konformacja wiązań z wyłączeniem trans do 13-cis. Model działania białka został stworzony w oparciu o badania z wykorzystaniem spektroskopii fourierowskiej oraz spektroskopii UV-VIS. INŻYNIERIA GENETYCZNA: Gen kodujący proteorodopsynę po przeniesieniu do komórki Escherichia coli umożliwiał zmodyfikowanej bakterii przenoszenie protonów przez błonę komórkową w obecności światła. Potwierdzono również, że gradient protonowy może być wykorzystany do syntezy ATP.
.png)
Źródło: 2011.igem.org

Źródło: Wikipedia. Proteorodopsyna.
|

Bardzo ciekawie napisane. Pozdrawiam serdecznie.
OdpowiedzUsuń