
 1) Destylacja - jest to rozdzielanie ciekłej mieszaniny wieloskładnikowej poprzez odparowanie, a następnie skroplenie jej składników. Stosuje się ją w celu wyizolowania lub oczyszczenia jednego lub więcej związków składowych. Proces wykorzystuje różną lotność względną składników mieszaniny. Główny produkt destylacji (czyli skroplona ciecz) nazywany jest destylatem. Pozostałość po destylacji nazywana jest cieczą wyczerpaną.
1) Destylacja - jest to rozdzielanie ciekłej mieszaniny wieloskładnikowej poprzez odparowanie, a następnie skroplenie jej składników. Stosuje się ją w celu wyizolowania lub oczyszczenia jednego lub więcej związków składowych. Proces wykorzystuje różną lotność względną składników mieszaniny. Główny produkt destylacji (czyli skroplona ciecz) nazywany jest destylatem. Pozostałość po destylacji nazywana jest cieczą wyczerpaną. |
| Źródło: Wikipedia. Alembik do destylacji. |
Najważniejszymi urządzeniami zwiększającymi efektywność destylacji są: kolumna rektyfikacyjna (kolumna destylacyjna) i deflegmator (który jest również rodzajem kolumny rektyfikacyjnej, o specyficznej konstrukcji i zastosowaniu).
RODZAJE DESTYLACJI:
1) Destylacja prosta (destylacja różniczkowa, destylacja kotłowa) - jest to destylacja polegająca na jednorazowym odparowaniu i skropleniu cieczy. Jest to proces okresowy, w którym para powstająca w czasie wrzenia cieczy, zawierająca więcej związków niskowrzących, jest natychmiast odprowadzana z przestrzeni parowania. Powoduje to stopniowe zmiany składu układu gaz-ciecz, czyli obu współistniejących faz. Wskutek zmian składu pary odbierane są frakcje destylatu o różnej temperaturze wrzenia. Metoda nie umożliwia odzyskania składników w postaci czystej. Jest stosowana w przypadkach, gdy ich składniki znacząco różnią się lotnością. Do grupy "destylacji prostych" bywa zaliczana destylacja równowagowa (rzutowa), która jest procesem ciągłym, ale prowadzi do rozdzielenia mieszaniny na tylko dwie frakcje o różnej lotności (wywar i destylat). Sprawność rozdzielania składników metodami destylacji kotłowej i rzutowej można zwiększyć stosując wielokrotne powtórzenia procesu (destylacja wielostopniowa), jednak bardziej korzystne jest ich zastępowanie ciągłymi procesami rektyfikacji. Fizykochemiczne podstawy: Efektywność rozdzielania mieszanin ciekłych (roztworów) zależy od względnych lotności ich składników, a pośrednio od wielkości sił oddziaływań międzycząsteczkowych w cieczy. Wpływa to na kształt i wielkość obszarów współistnienia w stanie równowagi pary i cieczy (szerokość tzw. "rybki" układów zeoptropowych), w tym na wielkość różnic między składem obu faz w różnych temperaturach, możliwość tworzenia azeotropów, heterozeotropów, heteroazeotropów lub innych układów o ograniczonej mieszalności cieczy.



W przypadku układów dwuskładnikowych AB przebieg destylacji może być przedstawiany z wykorzystaniem wykresów fazowych równowagi ciecz - para w układzie współrzędnych temperatura (T) – udział molowy (np. xB). Destylacja cieczy o określonej wartości xBrozpoczyna się w temperaturze, w której linia składu przecina linię temperatur wrzenia (dolna granica „rybki”). W tj temperaturze powstaje pierwszy pęcherzyk pary, która jest bogatsza od cieczy w składnik bardziej lotny (np. B) lub bardziej lotny dodatni azeotrop. Ten skład określa punkt przecięcia izotermy z góną granicą „rybki”, czyli krzywą pary nasyconej.

Źródło: Wikipedia. Laboratoryjny zestaw destylacyjny: 1 - palnik; 2 - kolba; 3 - nasadka destylacyjna; 4 - termometr; 5 - chłodnica; 6 - oliwka wlotowa wody; 7 - oliwka wylotowa wody; 8 - odbieralnik; 9 - oliwka próżniowa/gazu obojętnego; 10 - łącznik destylacyjny.
W chwili odprowadzenia z kolby do chłodnicy pierwszej porcji pary zaczyna się zmniejszać zawartość lotniejszego składnika (B) we wrzącej cieczy. Równocześnie wzrasta temperatura wrzenia roztworu. Zawartość B w kolejnych porcjach pary jest również coraz mniejsza. Prowadząc destylację można odpływające z chłodnicy porcje kondensatu (frakcje) kierować do odrębnych odbieralników, notując zakres temperatur wrzenia.

Źródło: Wikipedia. Laboratoryjny zestaw destylacyjny, umożliwiający odbieranie kolejnych frakcji: 1 - kolba, 2 - termometr, 3 - chłodnica, 4 - oliwka próżniowa, 5 - krówka destylacyjna, 6 - odbieralniki frakcji.
APARATURA: Destylacja prosta jest jednym z najstarszych procesów przeróbczych mających zastosowanie w alchemii. Naczynie do jej prowadzenia nazywano dawniej alembikiem. We współczesnych laboratoriach destylowana ciecz jest ogrzewana do wrzenia w kolbie z nasadką destylacyjną lub specjalnej kolbie destylacyjnej. Powstająca para nasycona jest kierowana stąd wprost do chłodnicy, w której skrapla się (kondensacja). W przemyśle stosowane są kotły i skraplacze o różnej konstrukcji. Kondensat, wypływający z chłodnicy w czasie destylacji w różnych zakresach temperatury wrzenia (frakcje), jest gromadzony w osobnych naczyniach. W laboratoriach odbieralniki są łączone z chłodnicą za pośrednictwem tzw. "krówki". Kondensaty dwufazowe (np. warstwa wodna i organiczna), znacznie różniące się chemicznym składem, są rozdzielany np. z użyciem rozdzielaczy laboratoryjnych lub przemysłowych separatorów, analogicznych do stosowanych w procesach ekstrakcji.
2) Destylacja równowagowa (inaczej rzutowa) - jest to rodzaj destylacji polegający na utrzymywaniu współistniejących faz, cieczy i pary, w stanie zbliżonym do równowagi, przy równoczesnym ciągłym dostarczaniu roztworu rozdzielanego (surówka) oraz odprowadzaniu odpowiednich ilości pary (destylat) i cieczy wyczerpanej (wywar). Destylacja równowagowa bywa zaliczana do grupy "destylacji prostych". Temperatura, która jest utrzymywana w destylatorze, mieści się między temperaturą wrzenia rozdzielanego roztworu (granica dolna), a temperaturą górną, w której - przy ogrzewaniu - odparowuje ostatnia kropla cieczy. Od temperatury, wybranej w tym zakresie, zależy skład strumieni rozdzielonych, odbieranych z urządzenia: cieczy i kondensatu, otrzymywanego po skropleniu pary (destylat). Surówka może być podgrzewana np. w wymienniku ciepła lub piecu rurowym. Jest wskazane stosowanie konstrukcji wymiennika zapewniającej jak najlepszy kontakt cieczy i powstającej pary. Mieszanina obu faz przepływa do separatora, z którego dolnej części odpływa wywar. Strumień pary jest kierowany do skraplacza i odbierany jako destylat. W urządzeniach do równowagowej destylacji ekspansywnej rozdzielany roztwór jest ogrzewany do temperatury niższej od temperatury wrzenia w aparacie o podwyższonym ciśnieniu. Jego rozprężanie w zaworze redukcyjnym powoduje samorzutne częściowe odparowanie. Strumień pary i cieczy jest kierowany do separatora obu faz, skąd para odpływa do skraplacza.


3) Destylacja wielostopniowa (destylacja wielokrotna) - jest to rzadko stosowany proces destylacji, polegający na kilkakrotnym powtórzeniu destylacji równowagowej lub prostej. Prowadzony jest w ten sposób, że destylat lub frakcja z jednego stopnia są kierowane na kolejny stopień jako roztwór do destylacji (roztwór surowy, surówka). Są przenoszone do następnej kolby destylacyjnej lub kierowane do następnego kotła. Ułatwia to wyodrębnienie określonych związków z ich mieszanin. Wielokrotne przenoszenie destylatu do kolejnych kotłów pozwala uzyskać końcowy destylat, który jest dużo bardziej wzbogacony w składnik bardziej lotny (np. B w układzie A+B), jednak jego ilość jest mała. Wydajność procesu jest tym mniejsza, im więcej zastosowano stopni destylacji, w celu uzyskania czystego produktu. Z każdego ze stopni odprowadzana jest "ciecz wyczerpana", w której zawartość składnika B zwiększa się ze stopnia na stopień. By poprawić efektywność wyodrębniania związki B w całym zestawie ciecz wyczerpaną z wszystkich kotłów poza pierwszym zawraca się do kotłów poprzednich (skład cieczy wyczerpanej z kotła n jest zwykle zbliżony do składu destylatu n-1). Uzyskuje się w ten sposób przepływ obu strumieni - cieczy i pary - w przeciwnych kierunkach. Jeżeli kotły są ustawione na coraz większej wysokości (schodkowo), przeciwprądowy przepływ jest samoczynny. Ciecz wyczerpana jest odprowadzana tylko z najniższego kotła zestawu destylatorów, a destylat - z najwyższego. Surówkę wprowadza się do jednego ze środkowych kotłów zestawu, dzięki czemu ilość składnika lotniejszego, która jest odprowadzana z cieczą wyczerpaną, jest mniejsza. Działanie opisanego zestawu do destylacji wielostopniowej wymaga dostarczania dużych ilości energii, potrzebnej do skroplenia kolejnych destylatów i ogrzewania cieczy w kolejnych kotłach. Optymalizację układu osiąga się kierując parę z każdego kotła n wprost do kotła n+1. Para przenosi strumień ciepła, potrzebnego do destylacji w wyższym kotle. W tak skonstruowanym zestawie destylatorów nagrzewnica jest instalowana tylko w najniższym kotle, a chłodnica - tylko za kotłem najwyższym.

 |
| Źródło: Wikipedia. Laboratoryjna kolumna rektyfikacyjna z płaszczem elektrycznym. |
5) Rektyfikacja (Destylacja frakcyjna/ Destylacja frakcjonowana) - jest to z fizycznego punktu widzenia destylacja kaskadowa (wielopoziomowa), w której każdy stopień procesu jest zasilany produktem (destylatem) poprzedniego; z punktu widzenia inżynierii procesowej rektyfikacja jest procesem jednostkowym, w którym mieszanina ciekła jest rozdzielana na frakcje o różnej (zwykle zbliżonej) lotności. Rektyfikacja w warunkach przemysłowych prowadzona jest w specjalnych kolumnach rektyfikacyjnych, zapewniających adiabatyczne (przemiana adiabatyczna to proces termodynamiczny, podczas którego izolowany układ nie nawiązuje wymiany ciepła, lecz całość energii jest dostarczana lub odbierana z niego jako praca) warunki procesu, chociaż czasami stosuje się kolumny z płaszczem chłodzącym lub grzejnym lub kolumny strefowe (na pewnym odcinku ogrzewane a na innym chłodzone), aby zapewnić ich maksymalną sprawność. Na dużą skalę rektyfikacja jest stosowana w rafineriach w przemyśle petrochemicznym (nazywana też rafineryjnym). W wyniku przerobu ropy naftowej otrzymuje się benzynę i inne paliwa, a także surowce dla przemysłu chemicznego. W przemyśle spirytusowym rektyfikacja spirytusu surowego umożliwiała uzyskanie czystego spirytusu rektyfikowanego. Zminiaturyzowane kolumny rektyfikacyjne stosuje się w laboratoriach chemicznych, gdyż rektyfikacja jest o wiele wydajniejsza niż destylacja prosta.
- Kolumna rektyfikacyjna - jest to sprzęt laboratoryjny, a także urządzenie przemysłowe, stosowane np. na wielką skalę w rafineriach ropy naftowej, w kształcie pionowej rury (lub walca), w której zachodzi kaskadowy, wielopoziomowy proces destylacji frakcyjnej, zwany rektyfikacją. ZASADA DZIAŁANIA: Na dole kolumny umieszcza się kolbę lub kadź (kocioł) z rektyfikowaną cieczą, a na jej górze głowicę lub deflegmator, których zadaniem jest częściowe zawracanie skraplanej cieczy (orosienia) z powrotem do kolumny, a częściowe kierowanie jej do odbieralnika. Kolumny rektyfikacyjne działają zawsze w układzie przeciwprądowym. Z góry do dołu spływa, pod wpływem grawitacji, skroplona ciecz, zaś od dołu do góry podążają opary destylowanej mieszaniny, wskutek czego znajdują się one w stałym kontakcie z cieczą. Wewnątrz kolumny występują elementy konstrukcyjne, które dodatkowo zwiększają powierzchnię kontaktu cieczy z oparami. W najprostszym przypadku są to półki z wąskimi otworami. W teorii rektyfikacji przyjmuje się, że na jednym elemencie (półce) następuje pojedynczy akt destylacji, czyli odparowanie i ponowne skroplenie cieczy. W praktyce laboratoryjnej kolumny półkowe są stosowane rzadko. Zamiast półek używa się kształtek o skomplikowanych kształtach, którymi wypełnia się kolumny, lub też stosuje się wypustki wychodzące ze ściany kolumny do jej wnętrza. Efektywność kolumn podaje się w jednostkach zwanych półkami teoretycznymi, które odpowiadają pojedynczym aktom odparowania i skroplenia w izotermicznych warunkach równowagowych. Jeśli kolumna ma na przykład 100 półek teoretycznych, oznacza to, że w jej wnętrzu następuje rozdział równy 100 aktom zwykłej destylacji równowagowej. Na efektywność działania kolumny, oprócz wypełnienia, mają też wpływ warunki, w jakich odbywa się rektyfikacja: ciśnienie, temperatura i stosunek cieczy zawracanej do odbieranej. Czym większy jest ten stosunek, tym lepszy rozdział. Jednak jego zwiększanie skutkuje też dłuższym czasem całego procesu i wzrostem jego kosztów. Najlepszy rozdział uzyskuje się przy zachowaniu identycznego ciśnienia i temperatury na całej długości kolumny. Temperaturę kontroluje się za pomocą tzw. płaszcza termicznego.
- GŁOWICA REKTYFIKACYJNA - jest to sprzęt laboratoryjny będący częścią aparatury do rektyfikacji lub destylacji. Spełnia on tę samą rolę, co zwykła nasadka destylacyjna i łącznik destylacyjny, tyle że umożliwia kontrolowanie tempa wypływu destylowanej cieczy z aparatury destylacyjnej. Część skroplonej cieczy jest bowiem zawracana do układu, a część jest kierowana do odbieralnika. Głowica rektyfikacyjna umożliwia też zwykle prostą wymianę odbieralników w trakcie destylacji pod zmniejszonym ciśnieniem bez konieczności jej przerywania, czy stosowania krówki destylacyjnej. Istnieje wiele typów konstrukcji głowic, wszystkie one działają jednak według jednej ogólnej zasady. Głowicę przyłącza się połączeniem szlifowym na szczycie kolumny rektyfikacyjnej lub podłącza się ją bezpośrednio do kolby z destylowaną substancją. Do drugiego szlifu przyłącza się odbieralnik. Bezpośrednio nad miejscem przyłączenia kolumny znajduje się najpierw miejsce na termometr a następnie chłodnica w kształcie palca z charakterystyczną "łezką" na samym końcu, która jest skierowana w stronę rurki odpływowej. Dzięki tej "łezce" skroplona ciecz spływa najpierw do rurki, a nie do kolumny. Skroplona ciecz może być jednak zawracana do kolumny poprzez zamykanie kranu odpływowego, którym jest przedzielona rurka odpływowa. Kran ten umożliwia regulację proporcji objętości cieczy wędrującej do odbieralnika i zawracanej do układu. Nad szlifem odbieralnika rozpoczyna się układ wyrównywania ciśnienia. Składa się on z dwóch kranów (z których jeden musi być trójdrożny), dwóch oliwek i rurki która łączy odbieralnik z górną częścią chłodnicy palcowej. W trakcie trwania destylacji próżniowej, kran odcinający 1 jest otwarty zaś kran odcinający 2 ustawia się w pozycji "połączenie góry z dołem". W momencie zmiany odbieralnika kran odcinający 1 na chwilę się zamyka, zaś kran odcinający 2 ustawia się w pozycji "połączenie dołu z odpowietrzeniem". Gdy destylacja jest przeprowadzana pod normalnym ciśnieniem, kran odcinający 1 jest otwarty cały czas, zaś kran odcinający 2 ustawia się w pozycji "wszystkie trzy otwory połączone razem". Oprócz głowic regulowanych ręcznie istnieją też głowice regulowane automatycznie. W głowicach tych występują zawory iglicowe, którymi steruje się elektrycznie za pomocą odpowiedniego panelu sterującego. Głowice automatyczne są często zintegrowane w jeden element z kolumnami i umożliwiają one bardzo precyzyjną kontrolę szybkości odbioru destylowanej cieczy.

- DEFLEGMATOR (Skraplacz częściowy) - jest to urządzenie działające podobnie do kolumny rektyfikacyjnej, zwiększające efektywność procesu destylacji (służącego rozdzielaniu mieszanin ciekłych). Deflegmator jest rodzajem chłodnicy, w której następuje częściowe skroplenie par opuszczających kolbę destylacyjną (naczynie z wrzącą cieczą) lub kolumnę rektyfikacyjną. Powstała w ten sposób flegma (kondensat o większym, w stosunku do par, stężeniu składnika mniej lotnego) jest zawracana do kolby (w przypadku destylacji prostej) lub na najwyższą półkę kolumny rektyfikacyjnej (w przypadku rektyfikacji). Nieskroplona para, wzbogacona w deflegmatorze w składnik bardziej lotny (inaczej mówiąc, niżej wrzący), jest odprowadzana do skraplacza jako destylat. Flegma (Orosienie - refluks, odciek, powrót) to część pary opuszczające kolumnę rektyfikacyjną, skroplona w deflegmatorze i zawracana na górę kolumny; pozostała część pary, po przejściu przez skraplacz jest odbierana jako destylat (rektyfikat). Orosienie, spływając w dół w przeciwprą∂zie do pary, wzbogaca ją w składnik bardziej lotny. W niektórych procesach rektyfikacyjnych do orosienia dodaje się tak zwanej cieczy surowej.
- Poniżej przedstawione zostały kolumny rektyfikacyjne rafinerii ropy naftowej (źródło: Wikipedia):


6) Destylacja azeotropowa - jest to jeden z rodzajów destylacji z użyciem czynnika rozdzielającego, zmieniającego względne lotności pierwotnych skłądników rozdzielanej mieszaniny. W przypadku destylacji azeotropowej czynnik rozdzielający tworzy ze składnikiem lub składnikami rozdzielanej mieszaniny taki azeotrop, że staje się możliwe rozdzielenie związków bliskowrzących lub tworzących inne azeotropy. Jest korzystne, jeżeli w postających układach azeotropowych występuje obszar ograniczonej mieszalności cieczy, co ułatwia rozdzielanie. Do grupy procesów destylacji z użyciem czynnika rozdzielającego należy również destylacja ekstrakcyjna i destylacja zeotropowa.
DESTYLACJA AZEOTROPOWA W LABORATORIUM: Destylacja azeotropowa jest stosowana w preparatyce organicznej w celu usuwania ze środowiska reakcji chemicznej części reagentów, np. produktów, których obecność hamuje reakcję, w celu oczyszczania produktów syntez lub oznaczania ich czystości (np. wilgotności). Przykłady:
 |
| Źródło: Wikipedia. Destylacja w kolbach z nasadkami azeotropowymi dla czynnika roz- -dzielającego (żółty) lżejszego i cięższego od wody (niebieski). |
- oznaczanie zawartości wody metodą Deana-Starka,
- osuszanie kwasu n-walerianowego, izowalerianowego, n-kapronowego i innych przez destylację z dodatkiem ok. 40% benzenu w stosunku do masy preparatu (odrzucany jest przedgon wrzący poniżej 100 stopni Celsjusza),
- osuszanie fruktozy metodą destylacji w układzie fruktoza-etanol-benzen: etap 1 - rozpuszczenie fruktozy w gorącym alkoholu i dodanie benzenu, etap 2 - destylacja azeotropu potrójnego benzen - etanol - woda (64 stopni Celsjusza), etap 3 - destylacja azeotropu benzen - etanol (69,3 stopnia Celsjusza), etap 4 - krystalizacja cukru z bezwodnego alkoholu; azeotropowa estryfikacja, np. reakcja alkoholu etylowego z kwasem szczawiowym, maleinowym lub benzoesowym w obecności takich czynników azeotropujących jak chloroform lub czterochlorek węgla.
DESTYLACJA AZEOTROPOWA W LABORATORIUM: Destylacja azeotropowa jest stosowana w preparatyce organicznej w celu usuwania ze środowiska reakcji chemicznej części reagentów, np. produktów, których obecność hamuje reakcję, w celu oczyszczania produktów syntez lub oznaczania ich czystości (np. wilgotności). Przykłady:
- oznaczanie zawartości wody metodą Deana - Starka,
- osuszanie kwasu n-walerianowego, izowalerianowego, n-kapronowego i innych przez destylację z dodatkiem ok. 40% benzenu w stosunku do masy preparatu (odrzucany jest przedgon wrzący poniżej 100 stopni Celsjusza); osuszanie fruktozy metodą destylacji w układzie fruktoza - etanol - benzen: etap 1 - rozpuszczenie fruktozy w gorącym alkoholu i dodanie benzenu, etap 2 - destylacja azeotropu potrójnego benzen - etanol - woda (64 stopnie Celsjusza), etap 3 - destylacja azeotropu benzen - etanol (69,3 stopnie Celsjusza), etap 4 - krystalizacja cukru z bezwodnego alkoholu; azeotropowa estryfikacja, np. reakcja alkoholu etylowego z kwasem szczawiowym, maleinowym lub benzoesowym w obecności takich czynników azeotropujących, jak chloroform lub czterochlorek węgla.

DESTYLACJA AZEOTROPOWA W PRZEMYŚLE: Rozdzielanie mieszanin metodą destylacji azeotropowej może być prowadzone w sposób ciągły, w zestawach kolumn rektyfikacyjnych, skraplaczy i rozdzielaczy dwufazowych kondensatów. Stosunkowo prosty przykład dotyczy zastosowania układu dwóch kolumn rektyfikacyjnych do rozdzielania związków A i B, które tworzą azeotrop dodatni, a ponadto charakteryzują się ograniczoną mieszalnością w temperaturach niższych od temperatury wrzenia azeotropu. Surówka A+B może być rozdzielana w kolumnie 1 na czysty składnik A (pierwsza ciecz wyczerpana) oraz pary o składzie zbliżonym do składu azeotropu AB. Są one kierowane do skraplacza, w którym kondensują z utworzeniem dwóch faz ciekłych. Faza, w której zawartość składnika A jest większa niż w parach z kolumny 1, jest zawracana do tej kolumny w celu odzyskania A. Faza druga zawiera mniej składnika A niż azeotrop. Może być rozdzielona w kolumnie 2 na czysty składnik B (druga ciecz wyczerpana) oraz pary o składzie zbliżonym do składu. Najlepiej znanym przykładem przemysłowej destylacji azeotropowej jest proces odwadniania azeotropu etanol - woda (95,57% C2H5OH) z użyciem benzenu jako czynnika rozdzielającego. Proces jest prowadzony w zestawie trzech kolumn rektyfikacyjnych. W kolumnie 1 jest oddestylowany trójskładnikowy azeotrop: etanol (18,5%) - woda (8,5%) - benzen (74%). Cieczą wyczerpaną jest bezwodny etanol (produkt). Po kondensacji par azeotropu potrójnego powstają dwie fazy: benzenowo - wodna (84,5,5 benzenu) i etanolowo-wodna (53% etanolu), zawierająca niewielką ilość benzenu. Roztwór benzenowy zawraca się do kolumny 1, a wodno-etanolowy kieruje się do kolumny 2, w której jest odpędzany benzen (jako azeotrop potrójny). Ciecz wyczerpaną z kolumny 2 - wodny roztwór etanolu - poddaje się destylacji w kolumnie 3, w której odzyskuje się etanol w formie podwójnego azeotropu z wodą, zawracanego do kolumny 1. Cieczą wyczerpaną jest woda.
 |
| Źródło: Wikipedia. Schemat odwadniania etanolu metodą destylacji azeotropowej z użyciem benzenu jako czynnika rozdzielającego: E - etanol, B - benzen, W - woda. |
7) Destylacja zeotropowa - jest to jeden z rodzajów destylacji z użyciem czynnika rozdzielającego, zmieniającego względne lotności składników rozdzielanej mieszaniny cieczy (surówka). W przypadku "destylacji zeotropowej" czynnik rozdzielający jest bardziej lotny od składników surówki i nie tworzy z nimi azeotropu (w przeciwieństwie do "destylacji ekstrakcyjnej" i "azeotropowej"). Jest odbierany ze szczytu kolumny wraz ze składnikiem lżejszym.

Przykład dotyczy rozdzielania dwuskładnikowej zeotropowej mieszaniny A + B, w której A jest składnikiem o niższej temperaturze wrzenia (bardziej lotnym). Rozdzielenie takich mieszanin jest trudne, gdy lotności obu składników są zbliżone. Niezbędne jest wówczas stosowanie wielopółkowych kolumn rektyfikacyjnych.
Do mieszaniny A + B wprowadza się trzeci składnik C, bardziej lotny od A, który w różnym stopniu zmienia lotności składników A i B. Od kształtu powierzchni pary nasyconej na wykresie fazowym temperatura - skład dla trójskładnikowego układu ciecz - para, zależy wielkość kolumn rektyfikacyjnych, które umożliwiają:
- etap 1 - oddzielenie mieszaniny lotnych składników A i C (dwuskładnikowy destylat) od mało lotnego B (ciecz wyczerpana z kolumny 1),
- etap 2 - rozdzielenie składników lotnych i odzyskanie czystego związku A (ciecz wyczerpana) i związku C (czynnik rozdzielający, zawracany do dolnej części kolumny 1 w postaci pary).
Rozdzielanie składników mieszanin metodą destylacji zeotropowej jest łatwiejsze w sytuacjach, gdy czynnik rozdzielający tworzy układy z obszarami współistnienia faz ciekłych (heterozeotropy lub obszary ograniczonej mieszalności cieczy poniżej powierzchni temperatur wrzenia).
8) Destylacja ekstrakcyjna - jest to jeden z rodzajów destylacji z użyciem czynnika rozdzielającego, czyli dodatkowego składnika, wprowadzanego do rozdzielanej mieszaniny w celu zmiany względnej lotności jej pierwotnych składników. Zwiększenie stosunku lotności umożliwia rozdzielenie mieszaniny lub ułatwia proces rozdzielania. W przypadku destylacji ekstrakcyjnej czynnik rozdzielający jest mało lotny. Opuszcza urządzenie do destylacji wraz z mniej lotnym składnikiem lub składnikami mieszaniny - z cieczą pozostającą po odpędzeniu składników lotnych (ciecz wyczerpana). Do grupy procesów destylacji z użyciem czynnika rozdzielającego należy również destylacja azeotropowa i destylacja zeotropowa. Proces destylacji ekstrakcyjnej może być prowadzony w sposób ciągły, w zestawie dwóch kolumn rektyfikacyjnych. Najprostszym przypadkiem jest rozdzielanie mieszaniny dwóch składników o podobnej lotności (A i B) z użyciem czynnika rozdzielającego (E, "ekstrahent"). Na środkowe półki kolumny pierwszej jest wprowadzana rozdzielana mieszanina A+B, a na górne półki - czynnik rozdzielający E. Ponieważ jest on trudno lotny, odparowuje w minimalnym stopniu, a powoduje zwiększenie lotności składnika A, którego pary wędrują na wyższe półki, a następnie do wylotowego skraplacza.


Mieszanina E+A+B, z rosnącą ilością B i malejącą ilością A, spływa w dół. Z dolnej części kolumny odprowadza się roztwór B w E, który wprowadza się na środkowe półki drugiej kolumny. Jest ona zwykle mniejsza od pierwszej, ponieważ różnice między lotnościami B i E są duże. Ten etap procesu pozwala odzyskać czysty składnik B oraz czynnik rozdzielający E, który jest zawracany na szczyt kolumny pierwszej. Ostateczny efekt przypomina ekstrakcję składnika B z mieszaniny A+B (pary płynące w górę) rozpuszczalnikiem E (ciecz spływający w dół), co wyjaśnia nazwę "destylacja ekstrakcyjna".
9) Destylacja z parą wodną - jest to destylacja z użyciem strumienia nasyconej lub przegrzanej pary wodnej, prowadząca do otrzymywania kondensatów dwufazowych (faza wodna i organiczna), odbieranych w temperaturze niższej od temperatury wrzenia najniżej wrzącego składnika mieszaniny (heteroazeotrop). Strumień pary wodnej jest doprowadzany do destylowanej mieszaniny z zewnątrz lub wytwarzany wewnątrz urządzenia do destylacji, na przykład przez intensywne ogrzewanie wodnego roztworu rozdzielanej mieszaniny związków chemicznych. Destylację z parą wodną stosuje się do wyodrębniania związków w niewielkim stopniu mieszających się z wodą (hydrofobowych), których destylacja prosta nie jest wskazana, głównie ze względu na możliwość termolizy. Metodą destylacji z parą wodną wyodrębnia się między innymi z surowców zielarskich pożądane składniki do leków ziołowych, a także olejków eterycznych (stosowanych do wytwarzania kosmetyków i przypraw oraz w aromaterapii). Metoda ta jest równie stosowana w preparatyce chemicznej do wyodrębniania produktów łatwo rozkładających się podczas syntezy z roztworów reakcyjnych oraz do rozdzielania skłądników mieszanin przy założeniu, że co najmniej jeden składnik destyluje z parą wodną.
Podstawy fizykochemiczne: Zgodnie z prawem Daltona całkowita prężność pary p nad cieczą dwuskładnikową jest sumą cząstkowych prężności par składników: p = pA + pB, które oblicza się na podstawie molowych udziałów (y) obu składników w pare: pA = p*yA; pB = p*yB. W przypadku roztworów doskonałych wartości pA i pB oblicza się na podstawie prawa Raoulta.
 |
| Źródło: Wikipedia. Francois Marie Raoult. |
 ,
,  - ciśnienie cząstkowe,
- ciśnienie cząstkowe,  - ciśnienie pary nasyconej czystej substancji ciekłej "i" w tej samej temperaturze,
- ciśnienie pary nasyconej czystej substancji ciekłej "i" w tej samej temperaturze,  - zawartość składnika "i" (ułamek molowy) w fazie ciekłej (indeks "I"). Prawo Raoulta pozwala określić ciśnienie pary nasyconej P mieszaniny o określonym składzie:
- zawartość składnika "i" (ułamek molowy) w fazie ciekłej (indeks "I"). Prawo Raoulta pozwala określić ciśnienie pary nasyconej P mieszaniny o określonym składzie: a także skład pary (indeks "v"), z reguły inny niż skład cieczy:
a także skład pary (indeks "v"), z reguły inny niż skład cieczy: . Zgodnie z prawem Raoulta faza gazowa (para cieczy) będzie zawsze bogatsza od fazy ciekłej w składniki łatwiej lotne (mające wyższe ciśnienie pary nasyconej a niższą temperaturę wrzenia).
. Zgodnie z prawem Raoulta faza gazowa (para cieczy) będzie zawsze bogatsza od fazy ciekłej w składniki łatwiej lotne (mające wyższe ciśnienie pary nasyconej a niższą temperaturę wrzenia).
ZASTOSOWANIA I ODCHYLENIA OD PRAWA RAOULTA: Prawo Raoulta jest spełnione w pełni dla roztworów idealnych. Roztwory rzeczywiste wykazują dobrą zgodność z prawem Raoulta, jeśli zawierają składniki o podobnym charakterze, np. benzen i toluen. W innych przypadkach prawo to jest spełnione jedynie dla dużych stężeń substancji rozpuszczonej; dla małych stężeń spełnione jest natomiast prawo Henry'ego, a dla stężeń pośrednich obserwuje się znaczące odstępstwa od odu praw.
Jeżeli ciśnienie pary nasyconej jest wyższe niż przewidywane przez prawo Raoulta, mówi się o dodatnim odchyleniu od prawa Raoulta. Jeśli ciśnienia pary nasyconej są mniejsze, wówczas mówi się o odchyleniach ujemnych. Jeżeli odchylenia są tak duże, że na wykresie P(x) pojawia się ekstremum (maksimum lub minimum) mówi się o azeotropach.
Prawo Raoulta pozwala wytłumaczyć podwyższenie temperatury wrzenia dla cieczy zawierających substancje nielotne - ciśnienie nad cieczą przy standardowej temperaturze wrzenia, określonej przez ciśnienie zewnętrzne jest niższe niż wymagane od wrzenia. Wymagane jest więc ogrzanie cieczy do wyższej temperatury. Różnica składu fazy ciekłej i gazowej umożliwia sotsowanie procesu destylacji do rozdzielania mieszanin ciekłych. W procesie tym faza gazowa (para) jest wzbogacana w składnik lotniejszy, a faza ciekła jest wzbogacana w składnik mniej lotny. Mieszaniny ciekłe posiadające punkt azeotropowy nie mogą być całkowicie rozdzielone na składniki w procesie destylacji.
 |
| Źródło: Wikipedia. Prężność par A i B nad roztworem doskonałym. |
 |
| Źródło: Wikipedia. Siły oddziaływań międzycząsteczkowych. |
 |
| Źródło: Wikipedia. Niewielkie dodatnie odchylenia od prawa Raoulta. |
 |
| Azeotrop dodatni aceton - CS2. |
 |
| Źródło: Wikipedia. Odchylenia od prawa Raoulta i ograniczona mieszalność cieczy. |
 |
| Źródło: Wikipedia. Heteroazeotrop. |
W przypadku roztworów niedoskonałych obserwowane są odchylenia od prawa Raoulta, których kierunek i wielkość zależą od wielkości sił oddziaływania między cząsteczkami A-A, B-B i A-B w cieczy (decydujących o wartościach współczynników aktywności). W przypadkach, gdy odchylenia od doskonałości są duże, w układach AB występują ekstremalne wartości prężności pary - maksymalne (azeotrop dodatni) lub minimalne (azeotrop ujemny). W przypadkach bardzo dużych odchyleń w układach występują obszary ograniczonej mieszalności cieczy (heteroazeotropia, heterozeotropia).
W przypadkach skrajnych dwie ciecze A i B, mieszające się w minimalnym stopniu, mogą być traktowane jak prawie niezależne układy ciecz-para. W każdym z nich w stałej temperaturze utrzymuje się taka sama prężność pary, niezależna od ilości obu cieczy Ogrzewane w odrębnych naczyniach zaczynają wrzeć w różnych temperaturach - takich, w których prężności par są równe wartości ciśnienia zewnętrznego. Jeżeli niemieszające się ciecze A i B znajdują się w jednym naczyniu, zależność prężności pary każdej z nich od temperatury nie zmienia się, ale cząsteczki cieczy dwufazowej prędzej ją opuszczają. Wrzenie rozpoczyna się w niższej temperaturze - wtedy, gdy wartość ciśnienia zewnętrznego jest osiągana przez sumę prężności A i B. W tak uproszczonym przypadku w stałej temperaturze skład pary nad roztworem jest niezależny od składu cieczy. Stosunek ilości moli A i B (nA i nB) w jednostce objętości pary jest równy stosunkowi odpowiednich prężności par (pA i pB) czystych składników w danej temperaturze. Stosunek mas obu składników (wA i wB) w parze jest zależny od prężności par czystych składników i ich ciężarów cząsteczkowych (MA i MB).


Efektywność odzyskiwania wysokowrzących związków chemicznych z ich mieszanin ze związkami nielotnymi z parą wodną można zwiększyć, stosując destylację z użyciem pary przegrzanej. Warunkiem ograniczającym są prawdopodobne reakcje rozkładu termicznego. Przykład: Benzaldehyd (tw. 178 stopni Celsjusza przy 760 mmHg) destyluje z równowagową parą wodną (100 stopni Celsjusza) w temperaturze 97,9 stopnia Celsjusza. Destylat zawiera 32,1% wag. benzaldehydu. Stosując parę wodną przegrzaną do 133 stopni Celsjusza otrzymuje się kondensat zawierający 70,6% wag. benzaldehydu.
SPRZĘT DO DESTYLACJI Z PARĄ WODNĄ: Instalacje do destylacji z parą wodną, niezależnie od ich skali, składają się z następujących elementów: wytwornicy pary, która bywa wyposażana w urządzenia do przegrzewania wylotowego strumienia lub zapobiegające porywaniu do destylatora kropel wody; zbiornik z materiałem zawierającym oddestylowany związek chemiczny, na przykład mieszaninę produktów syntezy organicznej lub surowcem zielarskim, rozłożonym na rusztach (destylator); chłodnicę (kondensator); rozdzielacz warstw dwufazowego kondensatu.






Standardowe zestawy laboratoryjne są montowane analogicznie do zestawów do destylacji prostej, z tym że kolba zamocowana jest pod kątem i nie musi być ogrzewana. Do kolby wprowadza się rurkę doprowadzającą strumień pary z centralnej sieci lub laboratoryjnej wytwornicy. Zamiast tradycyjnych korków z umieszczonymi rurkami, obecnie stosuje się specjalne szklane nasadki do destylacji z parą wodną. Tradycyjne metalowe kociołki, ogrzewane palnikami gazowymi, są wyposażone w poziomowskaz i wysoką rurkę, ograniczającą wahania ciśnienia i zapobiegającą wychlapywaniu wrzącej wody do strumienia pary. Po zakończeniu destylacji rurkę tę należy wysunąć ponad poziom cieczy lub usunąć z kociołka aby nie doszło do zassania cieczy z kolby do stygnącego kociołka. Jako wytwornice pary są też stosowane szklane kolby z elektrycznym płaszczem grzejnym również wyposażone w zabezpieczenie przed wahaniami ciśnienia.
Analogicznie działające urządzenia stosowano już w IV-III w p.n.e. w Grecji i Egipcie, w czasie wyodrębniania olejków eterycznych z surowców roślinnych. Znany do dzisiaj kocioł do destylacji, zwany alembikiem, jest prawdopodobnie wynalazkiem Arabów z VIII-IX w. n.e. (przypuszczalnie Dżabir Ibn Hajjan - znany jako "Geber").
Alembik przypomina warnik, stosowany przez dawnych gorzelników. Do jego owalnej kadzi jest mocowany kołpak zwany "łabędzią szyją", która pełni funkcję wstępnej powietrznej chłodnicy. Dalsze chłodzenie odbywa się w wężownicach umieszczonych w zbiornikach z wodą. Dwufazowy kondensat spływa do separatorów umożliwiających ciągłe odbieranie obu faz - np. olejku i wody kwiatowej.
PRZEBIEG DESTYLACJI Z PARĄ WODNĄ W LABORATORIUM: Strumień pary jest wprowadzany do kolby rurką sięgającą prawie do dna, często lekko zakrzywioną na końcu. Kolba jest montowana w takim położeniu, aby burzliwy przepływ pary przez ciecz nie powodował jej wychlapywania do chłodnicy. Strumień pary niosącej oddestylowane związki przepływa przez chłodnicę (wodną lub powietrzną), w której kondensują pary. Dwufazowy kondensat spływa do odbieralnika, którym może być np. standardowy rozdzielacz laboratoryjny albo urządzenia umożliwiające odbieranie obu faz w sposób ciągły, przez odpowiednio ulokowane rurki przelewowe. Oddestylowany składnik mieszaniny poddawanej destylacji (na przykład produkt prowadzonej w kolbie reakcji) może częściowo rozpuszczać się w wodzie (lub mieszać się z nią) co generuje straty. Aby uniknąć strat i zwiększyć wydajność procesu, warstwę wodną można w sposób ciągły zawracać do kolby destylacyjnej/reakcyjnej. W takim przypadku kolba musi być dodatkowo ogrzewana.
RODZAJE ZASTOSOWAŃ:
Wyodrębnianie produktów syntez organicznych: 1) Oddzielanie nieprzereagowanego aldehydu benzoesowego od kwasu cynamonowego, otrzymywanego w reakcji kondensacji tego aldehydu z bezwodnikiem octowym wobec octanu potasu (reakcja Perkina); 2) Oddzielenie głównego produktu jodowania benzenu (jodobenzenu) od produktów ubocznych; 3) Oddzielanie aniliny, otrzymywanej przez redukcję nitrobenzenu, od nieprzereagowanego substratu; 4) Rozdzielanie mieszaniny o-nitrofenolu i p-nitrofenolu, powstającej w czasie nitrowania fenolu. Różnica między prężnościami pary izomerów orto i para jest konsekwencją różnic w budowie cząsteczek. W przypadku izomeru orto tworzy się wewnątrzcząsteczkowe wiązanie wodorowe, które uniemożliwia asocjację cząsteczek. Asocjacji ulegają cząsteczki izomeru para, co powoduje zmniejszenie prężności pary. Ekstrakcja składników produktów naturalnych jest następująca: 1) Izolacja mentolu z mięty; 2) Izolacja olejku goździkowego. Utarte w moździerzu goździki (Eugenia caryophyllata) umieszcza się wraz z wodą w kolbie do destylacji z parą wodną. Zebrany kondensat jest poddawany dwukrotnej ekstrakcji chlorkiem metylenu. Z oddzielonej warstwy organicznej odparowuje się rozpuszczalnik, otrzymując olejek goździkowy, którego głównym składnikiem jest eugenol.
10) Destylacja pod zmniejszonym ciśnieniem (Destylacja próżniowa) - jest to destylacja prowadzona przy obniżonym ciśnieniu, zaliczana do zachowawczych metod rozdziału substancji. Dzięki zmniejszeniu ciśnienia możliwe jest obniżenie temperatury wrzenia cieczy, co ma istotne znaczenie, gdy któraś z wyodrębnianych substancji jest podatna na rozkład termiczny. Obniżenie ciśnienia wiąże się jednak ze zmniejszeniem się różnicy temperatur wrzenia składników rozdzielanej mieszaniny, dlatego w ten sposób rozdestylować można związki chemiczne o znacznej różnicy tych temperatur. Wyróżnia się:

W przypadku destylacji w warunkach wysokiej próżni lotności względne związków chemicznych zależą nie tylko od stosunku prężności par nasyconych składników destylowanej mieszaniny, lecz także od pierwiastka ze stosunku mas molowych rozdzielanych związków chemicznych (pod warunkiem, że nie występują między nimi wiązanie wodorowe i zbyt silne oddziaływania międzycząsteczkowe).
11) Destylacja cząsteczkowa (molekularna) - jest to rodzaj destylacji prowadzonej pod zmniejszonym ciśnieniem, charakteryzującej się stosowaniem wysokiej próżni (10−3–10−7 hPa) oraz bardzo małych odległości między strefą parowania cieczy i strefą skraplania pary, równych lub mniejszych od średniej drogi swobodnej cząsteczek. W tych warunkach cząsteczki, odparowujące z powierzchni cieczy (bez wrzenia), docierają bezpośrednio do skraplacza. W niesprężystych zderzeniach oddają zimnej powierzchni skraplacza swoją energię kinetyczną i zatrzymują się, tworząc warstewkę destylatu. Destylacja molekularna jest stosowana do wyodrębniania związków o wysokich temperaturach wrzenia, łatwo ulegających termolizie, np. koncentratów niektórych witamin i aromatów soków owocowych.
PODSTAWY FIZYKOCHEMICZNE: Podstawy destylacji molekularnej różnią się od podstaw innych rodzajów destylacji. W przypadku np. destylacji prostej, równowagowej, ekstrakcyjnej lub rektyfikacji rozdzielane roztwory doprowadza się do wrzenia. W czasie wrzenia para powstaje w całej objętości cieczy, co powoduje jej intensywne mieszanie. Powstaje para o zmieniającym się składzie, co pozwala odbierać frakcje lub pojedyncze składniki o różnych prężnościach pary nasyconej. Skład destylatów i cieczy wyczerpanej może być przewidywany w oparciu o wykresy fazowe równowagi między cieczą i parą. W przypadku destylacji molekularnej para powstaje wyłącznie na powierzchni. Jej prężność nie osiąga wartości ciśnienia zewnętrznego, więc nie dochodzi do wrzenia. Nie jest też osiągany stan równowagi ciecz-para. Składniki roztworu są rozdzielane w specyficznych warunkach, w których faza gazowa nie spełnia podstawowych kryteriów kinetyczno-molekularnej teorii gazów. Według tej teorii gaz jest ośrodkiem izotropowym. Składa się z chaotycznie poruszających się cząstek, które oddziałują na siebie poprzez zdęrzenia sprężyste. Drogą, jaką pokonują między zderzeniami (średnia droga swobodna), jest zależna od koncentracji (liczby cząsteczek w metrze sześciennym, n0) oraz tzw. efektywnej średnicy cząsteczek (d): .
 .
.
Wartość średniej drogi swobodnej jest podstawą definicji i klasyfikacji próżni w znaczeniu technicznym. W przypadku tzw. "próźni średniej" (1–10−3 hPa) λ ma wartość 0,1 mm – 10 cm, a w przypadku „próżni wysokiej” (HV; 10−3–10−7 hPa) – λ = 10 cm – 1 km.
W warunkach destylacji molekularnej faza gazowa nad parującą cieczą ma właściwości anizotropowe, ponieważ ruch cząsteczek nie jest chaotyczny. Tylko nieliczne spośród tych, które uwalniają się z powierzchni cieczy, zderzają się z innymi cząsteczkamif azy gazowej i - zmieniając kierunek ruchu - mogą wrócić na tę powierzchnię. Większość cząsteczek dociera bezpośrednio do przeciwległej powierzchni, która jest chłodzona (skraplacz). Tu oddają swoją energię kinetyczną i zostają zatrzymane (zderzenia niesprężyste). Sprawia to, że nie wzrasta ciśnienie parcjalne nad parującą cieczą i nie maleje szybkość parowania. Od powierzchni cieczy do skraplacza przepływa ukierunkowany strumień takich cząsteczek, których średnia droga swobodna jest podobna lub większa od odległości między oboma powierzchniami.
Zależność granicznej szybkości parowania (G [h/cm2*s]) od temperatury (T[K]), prężności pary (p [mmHg]) i masy molowej związku (M [g/mol]) wyraża równanie: . Prędkość rzeczywista jest mniejsza, ponieważ równanie nie uwzględnia możliwości powrotu części cząsteczek na parującą powierzchnię (np. parowania z powierzchni kondensacji). Mechanizm destylacji molekularnej znajduje istotne odbicie w specyficznym składzie destylatów, otrzymywanych w wyniku destylacji mieszanin. W przypadku klasycznej wysokotemperaturowej destylacji mieszaniny trzech związków liczby ich moli (N) mają się do siebie tak, jak odpowiednie prężności pary (p): . W przypadku destylacji molekularnej względne ilości składników w parze są zależne również od masy molowej: . Dzięki tej zależności destylacja molekularna ułatwia destylację związków wielkocząsteczkowych. W niektórych przypadkach jest jedyną możliwością oddestylowania związków o dużych cząsteczkach, które łatwo ulegają termolizie.
 . Prędkość rzeczywista jest mniejsza, ponieważ równanie nie uwzględnia możliwości powrotu części cząsteczek na parującą powierzchnię (np. parowania z powierzchni kondensacji). Mechanizm destylacji molekularnej znajduje istotne odbicie w specyficznym składzie destylatów, otrzymywanych w wyniku destylacji mieszanin. W przypadku klasycznej wysokotemperaturowej destylacji mieszaniny trzech związków liczby ich moli (N) mają się do siebie tak, jak odpowiednie prężności pary (p):
. Prędkość rzeczywista jest mniejsza, ponieważ równanie nie uwzględnia możliwości powrotu części cząsteczek na parującą powierzchnię (np. parowania z powierzchni kondensacji). Mechanizm destylacji molekularnej znajduje istotne odbicie w specyficznym składzie destylatów, otrzymywanych w wyniku destylacji mieszanin. W przypadku klasycznej wysokotemperaturowej destylacji mieszaniny trzech związków liczby ich moli (N) mają się do siebie tak, jak odpowiednie prężności pary (p):  . W przypadku destylacji molekularnej względne ilości składników w parze są zależne również od masy molowej:
. W przypadku destylacji molekularnej względne ilości składników w parze są zależne również od masy molowej:  . Dzięki tej zależności destylacja molekularna ułatwia destylację związków wielkocząsteczkowych. W niektórych przypadkach jest jedyną możliwością oddestylowania związków o dużych cząsteczkach, które łatwo ulegają termolizie.
. Dzięki tej zależności destylacja molekularna ułatwia destylację związków wielkocząsteczkowych. W niektórych przypadkach jest jedyną możliwością oddestylowania związków o dużych cząsteczkach, które łatwo ulegają termolizie. 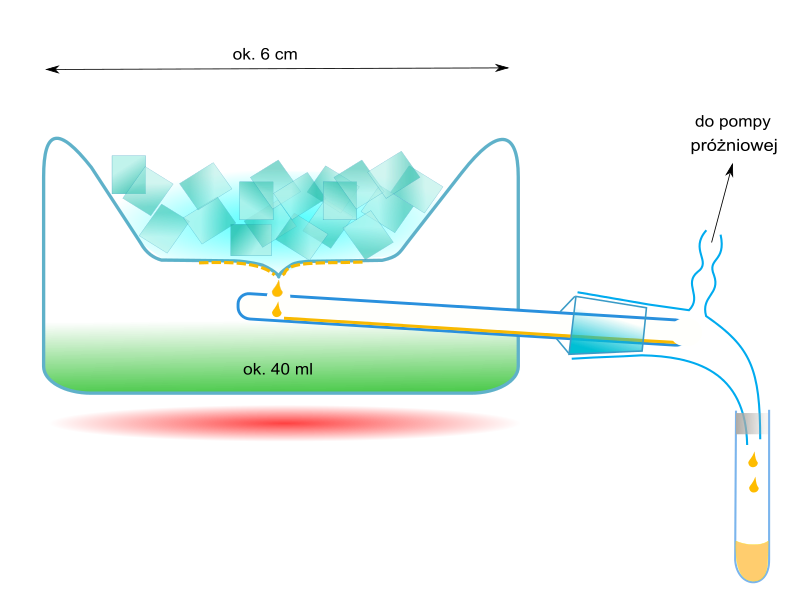 URZĄDZENIA DO DESTYLACJI MOLEKULARNEJ: Aparatura do destylacji molekularnej, stosowana w laboratoriach, np. w czasie wyodrębniania niewielkich ilości produktów syntez organicznych jest zróżnicowana. Przykładem urządzenia, stosowanego do destylacji ok. 40 cm3 roztworu, jest tzw. przyrząd destylacyjny Hickmana. Destylowana ciecz jest umieszczana w naczyniu o wymiarach ok. 6 x 4,5 cm, w którym jest wytwarzana próżnia. Aby wytworzyć odpowiednio wysoką próżnię stosuje się sprzęganie pomp olejowych, rotacyjnych i dyfuzyjnych (zwykle olejowych). W górnej ściance naczynia destylacyjnego znajduje się zagłębienie na mieszaninę chłodzącą. Na ochłodzonej ściance gromadzi się kondensat, który spływa do odbieralnika. W laboratoriach stosowane są również urządzenia o działaniu ciągłym, w którym surówka jest stopniowo dozowana na powierzchnię szklanej nagrzewnicy (np. zasilanej strumieniem ciepłej wody), po której spływa do odbieralnika cieczy wyczerpanej. Cząsteczki uwolnione z cieczy są zatrzymywane na wewnętrznej ściance naczynia próżniowego, w którym znajduje się nagrzewnica. Destylat spływa do odrębnego odbieralnika. Przykładem różnorodnych konstrukcji aparatów o działaniu ciągłym, stosowanych w skali przemysłowej, jest urządzenie z ogrzewanym wirnikiem w formie kielicha. Wewnątrz niego jest umieszczany drugi kielich, o chłodzonych ściankach (np. z płaszczem wodnym). Surówka jest wprowadzana na środek dna kielicha wirującego. Pod wpływem siły odśrodkowej przemieszcza się w kierunku brzegów, a następnie w górę, po nachylonych bocznych ściankach. Tworzy przy tym cienką warstewkę (film), dzięki czemu zwiększa się szybkość parowania (minimalizacja zahamowań, związanych z powolną dyfuzją z wnętrza cieczy do jej powierzchni).
URZĄDZENIA DO DESTYLACJI MOLEKULARNEJ: Aparatura do destylacji molekularnej, stosowana w laboratoriach, np. w czasie wyodrębniania niewielkich ilości produktów syntez organicznych jest zróżnicowana. Przykładem urządzenia, stosowanego do destylacji ok. 40 cm3 roztworu, jest tzw. przyrząd destylacyjny Hickmana. Destylowana ciecz jest umieszczana w naczyniu o wymiarach ok. 6 x 4,5 cm, w którym jest wytwarzana próżnia. Aby wytworzyć odpowiednio wysoką próżnię stosuje się sprzęganie pomp olejowych, rotacyjnych i dyfuzyjnych (zwykle olejowych). W górnej ściance naczynia destylacyjnego znajduje się zagłębienie na mieszaninę chłodzącą. Na ochłodzonej ściance gromadzi się kondensat, który spływa do odbieralnika. W laboratoriach stosowane są również urządzenia o działaniu ciągłym, w którym surówka jest stopniowo dozowana na powierzchnię szklanej nagrzewnicy (np. zasilanej strumieniem ciepłej wody), po której spływa do odbieralnika cieczy wyczerpanej. Cząsteczki uwolnione z cieczy są zatrzymywane na wewnętrznej ściance naczynia próżniowego, w którym znajduje się nagrzewnica. Destylat spływa do odrębnego odbieralnika. Przykładem różnorodnych konstrukcji aparatów o działaniu ciągłym, stosowanych w skali przemysłowej, jest urządzenie z ogrzewanym wirnikiem w formie kielicha. Wewnątrz niego jest umieszczany drugi kielich, o chłodzonych ściankach (np. z płaszczem wodnym). Surówka jest wprowadzana na środek dna kielicha wirującego. Pod wpływem siły odśrodkowej przemieszcza się w kierunku brzegów, a następnie w górę, po nachylonych bocznych ściankach. Tworzy przy tym cienką warstewkę (film), dzięki czemu zwiększa się szybkość parowania (minimalizacja zahamowań, związanych z powolną dyfuzją z wnętrza cieczy do jej powierzchni). 

Para kondensuje na zewnętrznej ściance zimnego kielicha, po której destylat spływa do otworów wylotowych, z których jest odpompowywany. Odpowiednie użebrowanie umożliwia odbieranie różnych frakcji, z różnych poziomów skraplacza. Analogiczna zasada, polegająca na rozprowadzaniu filmu cieczy na ruchomej powierzchni, jest wykorzystywana w innych typach urządzeń o działaniu ciągłym. Znane są instalacje, w których film cieczy jest wytwarzany na powierzchni obracającego się walca, położonego poziomo. Walec otacza skraplacz o kształcie cylindrycznym. Para kondensuje na wewnętrznej powierzchni cylindra i spływa do odpowiednio ukształtowanych rynienek.
WARUNKI POWODZENIA DESTYLACJI: Aby destylacja była skuteczna, skład gazu musi być różny od składu cieczy. Podstawowym warunkiem jest tu różnica temperatur wrzenia rozdzielanych związków chemicznych. Istnieją mieszaniny, których składników nie można rozdzielić przez "zwykłą" destylację, gdyż ciecze składowe podczas wrzenia wykazują identyczną prężność pary. Są to mieszaniny azeotropowe. Mieszaniny azeotropowe można rozdzielić metodą destylacji azeotropowej, która polega na rozdestylowaniu ich po uprzednim wprowadzeniu dodatkowego składnika, tzw. czynnika azeotropującego, który tworzy z substancjami azeotrop trójskładnikowy, wrzący w innej temperaturze i z innym stosunkiem składników niż mieszanina wyjściowa. Przykładem może być azeotrop etanol - woda (95,57% etanolu, temperatura wrzenia czystego etanolu 78,3 stopnia Celsjusza), który można rozdzielić po dodaniu pewnej ilości benzenu. Powstaje wówczas trójskładnikowa mieszanina azeotropowa - o składzie 7,5% wody, 18,5% etanolu, 74% benzenu - mająca temperaturę wrzenia 68,86 stopnia Celsjusza. Po całkowitym oddestylowaniu takiej mieszaniny pozostałością jest alkohol bezwodny (absolutny). Metoda ta była stosowana w przemyśle spirytusowym, obecnie nie jest ona stosowana ze względu na toksyczność benzenu.

1) Wymrażanie frakcyjne - jest to jeden z procesów stosowanych w inżynierii procesowej i chemii w celu rozdzielenia dwóch cieczy o różnych temperaturach krzepnięcia. Może być prowadzony przez częściowe topnienie fazy stałej lub przez częściową krystalizację fazy ciekłej. Krystalizacji tej można dokonać przez dodanie rozpuszczalnika do mieszaniny i schłodzenie roztworu lub odparowanie rozpuszczalnika. Wymrażanie frakcyjne ma również zastosowanie przy produkcji napojów alkoholowych w celu oczyszczenia wyrobu i zwiększenia zawartości alkoholu.
2) Rekrystalizacja frakcyjna - metoda oczyszczania krystalicznych związków chemicznych poprzez ich wielokrotne krystalizowanie i topienie bądź rozpuszczanie. Jedną z odmian rekrystalizacji jest krystalizacja strefowa.
 3) Sublimacja - jest to przemiana fazowa bezpośredniego przejścia ze stanu stałego w stan gazowy z pominięciem stanu ciekłego. Zjawisko odwrotne do sublimacji to resublimacja. Po raz pierwszy najdokładniej proces ten opisał nieznany alchemik średniowieczny zwany pseudo - Geberem (XII w.). W następnych wiekach sublimacja stała się procesem powszechnie stosowanym w laboratoriach chemicznych. Sublimację wykorzystuje się w laboratoriach i w przemyśle do oczyszczania względnie wydzielania z mieszanin jodu, kamfory, salmiaku (chlorku amonu), naftalenu, antracenu i innych substancji. Większość związków chemicznych nie sublimuje w temperaturze i przy ciśnieniu zbliżonym do warunków normalnych. Dla danej substancji sublimacja zachodzi w takich warunkach termodynamicznych (ciśnienie i temperatura), w których nie może ona istnieć w formie ciekłej. Warunki te określa ciśnienie punktu potrójnego dla tej substancji. Aby zachodziła sublimacja, ciśnienie parcjalne bezpośrednio nad powierzchnią sublimującego ciała musi być niższe od ciśnienia równowagi dla danej temperatury ciała. Dla ciśnień powyżej ciśnienia równowagi zachodzi resublimacja. Dla temperatur poniżej punktu potrójnego stan równowagi wyznacza zależność ciśnienia sublimacji od temperatury. Ze wzrostem ciśnienia wzrasta temperatura sublimacji, jednak jest ona zawsze niższa od temperatury punktu potrójnego. Przykładem ciała sublimującego w warunkach normalnych jest suchy lód czyli zestalony dwutlenek węgla. Jego punkt potrójny wypada przy ciśnieniu 5,1 atm i temperaturze -56,6 stopni Celsjusza, zatem blok zamrożonego dwutlenku węgla wystawiony do pomieszczenia, w którym panuje typowe ciśnienie atmosferyczne (ok. 1 atm) i temperatura ok. 20 stopni Celsjusza będzie sublimował, a nie topił się. Temperatura sublimacji suchego lodu przy ciśnieniu normalnym jest równa -78 stopni Celsjusza. Uzyskanie równowagi między cieczą a gazem dla dwutlenku węgla wymaga uzyskania ciśnienia powyżej 5,1 atm. Sublimacja jest często wykorzystywana do oczyszczania związków chemicznych, które mają na tyle wysoką temperaturę wrzenia, że ich destylowanie byłoby bardzo kłopotliwe. Ze względu na to, że większość związków organicznych ma punkt potrójny przy ciśnieniu znacznie niższym od ciśnienia atmosferycznego, sublimacja wymaga zwykle stosowania aparatury próżniowej.
3) Sublimacja - jest to przemiana fazowa bezpośredniego przejścia ze stanu stałego w stan gazowy z pominięciem stanu ciekłego. Zjawisko odwrotne do sublimacji to resublimacja. Po raz pierwszy najdokładniej proces ten opisał nieznany alchemik średniowieczny zwany pseudo - Geberem (XII w.). W następnych wiekach sublimacja stała się procesem powszechnie stosowanym w laboratoriach chemicznych. Sublimację wykorzystuje się w laboratoriach i w przemyśle do oczyszczania względnie wydzielania z mieszanin jodu, kamfory, salmiaku (chlorku amonu), naftalenu, antracenu i innych substancji. Większość związków chemicznych nie sublimuje w temperaturze i przy ciśnieniu zbliżonym do warunków normalnych. Dla danej substancji sublimacja zachodzi w takich warunkach termodynamicznych (ciśnienie i temperatura), w których nie może ona istnieć w formie ciekłej. Warunki te określa ciśnienie punktu potrójnego dla tej substancji. Aby zachodziła sublimacja, ciśnienie parcjalne bezpośrednio nad powierzchnią sublimującego ciała musi być niższe od ciśnienia równowagi dla danej temperatury ciała. Dla ciśnień powyżej ciśnienia równowagi zachodzi resublimacja. Dla temperatur poniżej punktu potrójnego stan równowagi wyznacza zależność ciśnienia sublimacji od temperatury. Ze wzrostem ciśnienia wzrasta temperatura sublimacji, jednak jest ona zawsze niższa od temperatury punktu potrójnego. Przykładem ciała sublimującego w warunkach normalnych jest suchy lód czyli zestalony dwutlenek węgla. Jego punkt potrójny wypada przy ciśnieniu 5,1 atm i temperaturze -56,6 stopni Celsjusza, zatem blok zamrożonego dwutlenku węgla wystawiony do pomieszczenia, w którym panuje typowe ciśnienie atmosferyczne (ok. 1 atm) i temperatura ok. 20 stopni Celsjusza będzie sublimował, a nie topił się. Temperatura sublimacji suchego lodu przy ciśnieniu normalnym jest równa -78 stopni Celsjusza. Uzyskanie równowagi między cieczą a gazem dla dwutlenku węgla wymaga uzyskania ciśnienia powyżej 5,1 atm. Sublimacja jest często wykorzystywana do oczyszczania związków chemicznych, które mają na tyle wysoką temperaturę wrzenia, że ich destylowanie byłoby bardzo kłopotliwe. Ze względu na to, że większość związków organicznych ma punkt potrójny przy ciśnieniu znacznie niższym od ciśnienia atmosferycznego, sublimacja wymaga zwykle stosowania aparatury próżniowej.  |
| Źródło: Wikipedia. Pary sublimującego jodu. |
 Poniżej przedstawiony został wykres fazowy z oznaczonym zakresem sublimacji (czerwona strzałka); p.p - punkt potrójny; p.k. - punkt krytyczny.
Poniżej przedstawiony został wykres fazowy z oznaczonym zakresem sublimacji (czerwona strzałka); p.p - punkt potrójny; p.k. - punkt krytyczny.
4) Resublimacja (desublimacja) - jest to przemiana fazowa polegająca na bezpośrednim przechodzeniu substancji z fazy gazowej (pary) w fazę stałą z pominięciem stanu ciekłego. Resublimacja jest procesem odwrotnym do sublimacji. W wyniku resublimacji wody (pary wodnej) powstaje szron i śnieg. Resublimacja, w połączeniu z sublimacją lub parowaniem, jest wykorzystywana do oczyszczania lub rozdzielania substancji i otrzymywania ich w postaci krystalicznej (jest to m.in. metoda oczyszczania jodu).
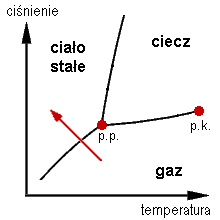 Poniżej przedstawiony został wykres fazowy z oznaczonym zakresem resublimacji (czerwona strzałka); p.p. - punkt potrójny; p.k. - punkt krytyczny.
Poniżej przedstawiony został wykres fazowy z oznaczonym zakresem resublimacji (czerwona strzałka); p.p. - punkt potrójny; p.k. - punkt krytyczny.  |
Źródło: Wikipedia. Para wodna po resublimacji na szybie, czyli szron. Źródło: Wikipedia. Resublimowany jod. |
5) Piroliza (gr. pyro, ogień i lysis, rozpad)/ sucha destylacja/ destylacja rozkładowa - jest to proces degradacji zachodzący pod wpływem wysokiej temperatury i prowadzony bez dostępu tlenu i innych celowo dodawanych reagentów. Podobnym procesem jest przebiegające w niższej temperaturze wytlewanie.



Poniższy film przedstawia proste urządzenie do prowadzenia procesu pirolizy:
INNE TERMINY ZWIĄZANE Z PROCESEM DESTYLACJI:
 1) Aparat Derynga - jest to powstałe w drugiej połowie lat 40. XX wieku laboratoryjne urządzenie do określania zawartości olejków eterycznych w materiale roślinnym, np. w surowcach roślinnych dla perfumerii i ziołolecznictwa, metodą destylacji z parą wodną. Konstrukcja aparatu została opracowana i opatentowana przez Jakuba Derynga, profesora na Wydziale Farmaceutycznym Akademii Medycznej w Warszawie. Aparat i procedurę pomiarów opisano w normach, polskich i międzynarodowych, a także w farmakopeach i podręcznikach akademickich. W czasie pomiaru zawartości olejków stosowane są kolby o pojemności 1000 lub 500 centymetrów sześciennych, w których jest umieszczana znana ilość badanego materiału i woda. Kolbę łączy się z zasadniczą częścią aparatu, której elementami są: kolumna destylacyjna, chłodnica (o różnej konstrukcji, zależnie od typu aparatu), wyskalowany odbieralnik (napełniany wodą przed rozpoczęciem destylacji). Odbieralnik jest połączony rurką przelewową z kolumną destylacyjną. Zawartość kolby jest utrzymywana w stanie wrzenia. Para wodna, wraz z porywanymi parami olejków, przepływa do chłodnicy, w której powstaje kondensat, rozwarstwiający się na dwie fazy. Dolna faza wodna jest zawracana do kolby. Olejek gromadzi się na powierzchni wody. Proces jest prowadzony przez ok. 3 godziny (zgodnie z przepisem, dotyczącym konkretnego badanego materiału). Po zakończeniu destylacji i ochłodzeniu aparatu olejek sprowadza się do wyskalowanej części odbieralnika i odczytuje jego objętość. Wynik jest przeliczany na 100 g surowca.
1) Aparat Derynga - jest to powstałe w drugiej połowie lat 40. XX wieku laboratoryjne urządzenie do określania zawartości olejków eterycznych w materiale roślinnym, np. w surowcach roślinnych dla perfumerii i ziołolecznictwa, metodą destylacji z parą wodną. Konstrukcja aparatu została opracowana i opatentowana przez Jakuba Derynga, profesora na Wydziale Farmaceutycznym Akademii Medycznej w Warszawie. Aparat i procedurę pomiarów opisano w normach, polskich i międzynarodowych, a także w farmakopeach i podręcznikach akademickich. W czasie pomiaru zawartości olejków stosowane są kolby o pojemności 1000 lub 500 centymetrów sześciennych, w których jest umieszczana znana ilość badanego materiału i woda. Kolbę łączy się z zasadniczą częścią aparatu, której elementami są: kolumna destylacyjna, chłodnica (o różnej konstrukcji, zależnie od typu aparatu), wyskalowany odbieralnik (napełniany wodą przed rozpoczęciem destylacji). Odbieralnik jest połączony rurką przelewową z kolumną destylacyjną. Zawartość kolby jest utrzymywana w stanie wrzenia. Para wodna, wraz z porywanymi parami olejków, przepływa do chłodnicy, w której powstaje kondensat, rozwarstwiający się na dwie fazy. Dolna faza wodna jest zawracana do kolby. Olejek gromadzi się na powierzchni wody. Proces jest prowadzony przez ok. 3 godziny (zgodnie z przepisem, dotyczącym konkretnego badanego materiału). Po zakończeniu destylacji i ochłodzeniu aparatu olejek sprowadza się do wyskalowanej części odbieralnika i odczytuje jego objętość. Wynik jest przeliczany na 100 g surowca.
APARAT W CZASIE I PO DESTYLACJI:

Druga z popularnych metod oznaczania zawartości olejków polega na stosowaniu procesu ekstrakcji, np. z użyciem aparatu Soxhleta.
2) Azeotrop - mieszanina azeotropowa - jest to ciekła mieszanina (roztwór) dwóch lub więcej związków chemicznych, która jest w równowadze termodynamicznej z parą nasyconą powstającą z tej mieszaniny. Skład pary i cieczy jest taki sam. Gdy mówimy o azeotropie w kontekście wykresu fazowego, parametry takie określamy nazwą punktu azeotropowego. Proporcje molowe (stężenie) związków chemicznych obecnych w parze nasyconej, powstającej w trakcie parowania mieszaniny azeotropowej, są takie, jak w samej cieczy. Oznacza to, że po skropleniu pary znad mieszaniny azeotropowej uzyskuje się ciecz o takim samym składzie chemicznym, jak wyjściowa mieszanina, co uniemożliwia jej rozdzielenie przez destylację, a nawet rektyfikację.
Mieszaniny azeotropowe rozdziela się zazwyczaj przez ekstrakcję, sorpcję lub poprzez dodanie do układu jeszcze jednego związku chemicznego, który tworzy azeotrop z jednym ze skłądników wcześniejszej mieszaniny i umożliwia dzięki temu wydestylowanie potrzebnego składnika. Klasycznym przykładem azeotropu jest spirytus rektyfikowany, czyli mieszanina wody z etanolem o stężeniu alkoholu (zależnie od temperatury i ciśnienia) od 95,5% do 97,5%. Aby uzyskać prawie czysty etanol, do azeotropu można dodać benzen i kontynuować rektyfikację. Pozwala to uzyskać roztwór etanolu zawierający około 99,8% alkoholu i niewielkie ilości benzenu. Obecnie, ze względu na toksyczność benzenu, tej metody już się nie stosuje. Zamiast niej stosowana jest destylacja pod ciśnieniem dwóch atmosfer, gdyż pod tym ciśnieniem etanol nie tworzy z wodą azeotropu.
 |
| Źródło: Azeotrop dodatni. Izobara i Izoterma równowagi fazowej między cieczą i parą. |
Mieszaniny azeotropowe rozdziela się zazwyczaj przez ekstrakcję, sorpcję lub poprzez dodanie do układu jeszcze jednego związku chemicznego, który tworzy azeotrop z jednym ze skłądników wcześniejszej mieszaniny i umożliwia dzięki temu wydestylowanie potrzebnego składnika. Klasycznym przykładem azeotropu jest spirytus rektyfikowany, czyli mieszanina wody z etanolem o stężeniu alkoholu (zależnie od temperatury i ciśnienia) od 95,5% do 97,5%. Aby uzyskać prawie czysty etanol, do azeotropu można dodać benzen i kontynuować rektyfikację. Pozwala to uzyskać roztwór etanolu zawierający około 99,8% alkoholu i niewielkie ilości benzenu. Obecnie, ze względu na toksyczność benzenu, tej metody już się nie stosuje. Zamiast niej stosowana jest destylacja pod ciśnieniem dwóch atmosfer, gdyż pod tym ciśnieniem etanol nie tworzy z wodą azeotropu.
 AZEOTROPIA: Azeotropia zachodzi w sytuacji gdy roztwór rzeczywisty wykazuje znaczne odstępstwa od prawa Raoulta. W takim kontekście można podzielić azeotropię na: dodatnią - następuje duże dodatnie odstępstwo od prawa Raoulta, a całkowita prężność pary nad mieszaniną jest większa odprężności pary czystego, lotniejszego składnika dla danej temperatury. Powstaje azeotrop dodatni; Ujemną - następuje duże ujemne odstępstwo od prawa Raoulta, a całkowita prężność pary nad mieszaniną jest mniejsza od prężności pary czystego, mniej lotnego składnika dla danej temperatury. Powstaje azeotrop ujemny. Azeotropia dodatnia jest częstsza od azeotropii ujemnej. Przykładami azeotropii dodatniej są: woda - etanol, benzen - metanol, benzen - etanol, benzen - tetrachlorek węgla, metanol - tetrachlorek węgla. Azeotropami ujemnymi są na przykład: aceton - chloroform, woda - kwas azotowy, woda - chlorowodór.
AZEOTROPIA: Azeotropia zachodzi w sytuacji gdy roztwór rzeczywisty wykazuje znaczne odstępstwa od prawa Raoulta. W takim kontekście można podzielić azeotropię na: dodatnią - następuje duże dodatnie odstępstwo od prawa Raoulta, a całkowita prężność pary nad mieszaniną jest większa odprężności pary czystego, lotniejszego składnika dla danej temperatury. Powstaje azeotrop dodatni; Ujemną - następuje duże ujemne odstępstwo od prawa Raoulta, a całkowita prężność pary nad mieszaniną jest mniejsza od prężności pary czystego, mniej lotnego składnika dla danej temperatury. Powstaje azeotrop ujemny. Azeotropia dodatnia jest częstsza od azeotropii ujemnej. Przykładami azeotropii dodatniej są: woda - etanol, benzen - metanol, benzen - etanol, benzen - tetrachlorek węgla, metanol - tetrachlorek węgla. Azeotropami ujemnymi są na przykład: aceton - chloroform, woda - kwas azotowy, woda - chlorowodór. 

4) Krówka destylacyjna - jest to sprzęt laboratoryjny służący do prowadzenia destylacji próżniowej. Łączy chłodnicę z kilkoma kolbkami lub probówkami, w które zbiera się różne frakcje destylowanej cieczy. Krówka ma kształt szklanego stożka z jedną szyjką u nasady i dwoma, trzema lub czterema szyjkami w podstawie. Szyjka u nasady jest zaopatrzona w szlif żeński, zaś szyjki u podstawy zakończone są szlifami męskimi. Krówkę montuje się górną szyjką do chłodnicy, zaś do dolnych szyjek przymocowuje się małe kolbki lub probówki ze szlifem, zwane odbieralnikami.

ZASADA DZIAŁANIA: W trakcie destylacji próżniowej wewnątrz aparatury musi być przez cały czas utrzymywane odpowiednie, niskie ciśnienie. Wymiana kolbek odbieralnikowych przy zastosowaniu zwykłego łącznika destylacyjnego, posiadającego tylko jeden wylot, jest niemożliwa bez rozszczelnienia całej aparatury i chwilowego przerwania procesu. Aby tego uniknąć i móc prowadzić go w sposób ciągły, wymyślono właśnie krówkę. Podczas destylacji krówkę można dowolnie obracać względem chłodnicy, wskutek czego kapiąca z niej ciecz wpada do wybranych odbieralników. Dzięki temu do kolejnych naczyń można w prosty sposób zbierać kolejne frakcje destylatu. Podobnie jak w trakcie każdej destylacji, o zmianie frakcji wnioskuje się na podstawie skokowej zmiany temperatury par wydobywających się z kolby, w której znajduje się destylowana mieszanina.
 5) Łącznik destylacyjny (przedłużacz) - jest to sprzęt laboratoryjny stosowany w trakcie prowadzenia destylacji, który służy do łączenia chłodnicy z kolbą, w której zbiera się destylowana ciecz. Łącznik destylacyjny ma kształt wygiętej rurki, w którą od jednej strony wtopiona jest druga rurka o mniejszej średnicy. Od strony, z której łączy się z chłodnicą, łącznik posiada zwykle szlif żeński, zaś od drugiej zakończony jest szlifem męskim. Oprócz tego w bocznej ściance, poniżej miejsca wtopienia wewnętrznej rurki, ale powyżej szlifu męskiego znajduje się oliwka [oliwka to specjalnie ukształtowana końcówka rurek szklanych, metalowych lub z tworzywa sztucznego, lub w rozmaitych sprzętach laboratoryjnych. Ich zadaniem jest zapewnienie szczelnego połączenia węży z aparaturą i zapobieganie ześlizgiwaniu się ich z króćców w trakcie pracy; króciec zaś to krótka rurka lub rura przymocowana (wspawana, wtopiona, przykręcona) do naczynia, zbiornika, aparatu lub urządzenia, do której przyłącza się wąż lub rurę doprowadzającą bądź odprowadzającą substancję lub czynnik. Króciec umożliwia też wprowadzanie do aparatu różnych narzędzi i przyrządów]. ZASADA DZIAŁANIA: Łącznik może służyć zarówno do destylacji atmosferycznej, jak i w atmosferze gazu obojętnego, a także do prostej destylacji próżniowej. We wszystkich tych przypadkach skroplona ciecz spływa z chłodnicy do łącznika, a następnie jest odprowadzana przez wewnętrzną rurkę do odbieralnika. Skręcony kształt łącznika ułatwia montowanie całej aparatury. Oliwka w bocznej ściance, w trakcie destylacji atmosferycznej służy jako odpowietrzenie całego układu destylacyjnego. W trakcie destylacji w atmosferze gazu obojętnego podłącza się do niej wąż połączony z systemem dostarczania tego gazu. W trakcie destylacji próżniowej do oliwki przyłącza się wąż od pompy próżniowej. Dzięki temu, że oliwka w bocznej ściance łącznika znajduje się znacznie powyżej wylotu wewnętrznej rurki, unika się zjawiska nadmiernego zasysania powietrza lub gazu obojętnego w czasie destylacji pod normalnym ciśnieniem, oraz unika się ryzyka zasysania cieczy do pompy próżniowej w czasie destylacji pod zmniejszonym ciśnieniem. W przypadku destylacji próżniowej, w której może występować wiele frakcji cieczy, praktyczniej jest jednak stosować krówkę lub głowicę.
5) Łącznik destylacyjny (przedłużacz) - jest to sprzęt laboratoryjny stosowany w trakcie prowadzenia destylacji, który służy do łączenia chłodnicy z kolbą, w której zbiera się destylowana ciecz. Łącznik destylacyjny ma kształt wygiętej rurki, w którą od jednej strony wtopiona jest druga rurka o mniejszej średnicy. Od strony, z której łączy się z chłodnicą, łącznik posiada zwykle szlif żeński, zaś od drugiej zakończony jest szlifem męskim. Oprócz tego w bocznej ściance, poniżej miejsca wtopienia wewnętrznej rurki, ale powyżej szlifu męskiego znajduje się oliwka [oliwka to specjalnie ukształtowana końcówka rurek szklanych, metalowych lub z tworzywa sztucznego, lub w rozmaitych sprzętach laboratoryjnych. Ich zadaniem jest zapewnienie szczelnego połączenia węży z aparaturą i zapobieganie ześlizgiwaniu się ich z króćców w trakcie pracy; króciec zaś to krótka rurka lub rura przymocowana (wspawana, wtopiona, przykręcona) do naczynia, zbiornika, aparatu lub urządzenia, do której przyłącza się wąż lub rurę doprowadzającą bądź odprowadzającą substancję lub czynnik. Króciec umożliwia też wprowadzanie do aparatu różnych narzędzi i przyrządów]. ZASADA DZIAŁANIA: Łącznik może służyć zarówno do destylacji atmosferycznej, jak i w atmosferze gazu obojętnego, a także do prostej destylacji próżniowej. We wszystkich tych przypadkach skroplona ciecz spływa z chłodnicy do łącznika, a następnie jest odprowadzana przez wewnętrzną rurkę do odbieralnika. Skręcony kształt łącznika ułatwia montowanie całej aparatury. Oliwka w bocznej ściance, w trakcie destylacji atmosferycznej służy jako odpowietrzenie całego układu destylacyjnego. W trakcie destylacji w atmosferze gazu obojętnego podłącza się do niej wąż połączony z systemem dostarczania tego gazu. W trakcie destylacji próżniowej do oliwki przyłącza się wąż od pompy próżniowej. Dzięki temu, że oliwka w bocznej ściance łącznika znajduje się znacznie powyżej wylotu wewnętrznej rurki, unika się zjawiska nadmiernego zasysania powietrza lub gazu obojętnego w czasie destylacji pod normalnym ciśnieniem, oraz unika się ryzyka zasysania cieczy do pompy próżniowej w czasie destylacji pod zmniejszonym ciśnieniem. W przypadku destylacji próżniowej, w której może występować wiele frakcji cieczy, praktyczniej jest jednak stosować krówkę lub głowicę. 

 |
| Źródło: Wikipedia. Dane do określania liczby stopni kolumny rektyfikacyjnej metodą McCabe'a i Thielego oraz wynik zastosowania metody.
PRZEBIEG WYZNACZANIA LICZBY PÓŁEK TEORETYCZNYCH: Przed rozpoczęciem oznaczenia liczby półek teoretycznych w określonej kolumnie rektyfikacyjnej trzeba uzyskać dane dotyczące równowagi ciecz-para w testowym układzie dwuskładnikowym A + B (B - składnik bardziej lotny). W czasie rektyfikacji roztworu testowego oznacza się wielkości strumieni destylatu i orosienia (liczba moli na godzinę) oraz stężenia składników w destylacie, surówce i cieczy wyczerpanej (ułamki molowe).
Sporządzany jest wykres w układzie współrzędnych: oś odciętych - x, ułamek molowy B w cieczy, oś rzędnych -y - ułamek molowy B w parze. W pierwszej kolejności wykreśla się linię pomocniczą x = y (przekątna kwadratu), linię równowagi - zależność ułamka molowego B w parze od ułamka molowego B w cieczy; linie pionowe, określające ułamek molowy B w surówce, destylacie i cieczy wyczerpanej. W następnej kolejności wykreśla się tzw. górną linię operacyjną, dla tej części kolumny, która znajduje się nad punktem odpowiadającym składowi surówki (większe stężenie składnika bardziej lotnego). W tym celu z punktu przecięcia linii składu destylatu z linią pomocniczą x = y wykreśla się prostą o nachyleniu:
gdzie:
Przykład:
W kolejnym etapie jest wykreślana tak zwana „linia q”. Parametr q określa termodynamiczny stan surówki. Jest zdefiniowany jako ułamek molowy cieczy w surówce, która może być np. mieszaniną cieczy i pary, parą nasyconą bez cieczy lub cieczą o temperaturze niższej od temperatury wrzenia.
Współczynnik nachylenia „linii q” oblicza się jako:
Przypadki charakterystyczne:
W przypadkach, gdy surowiec jest przegrzaną parą lub ochłodzoną cieczą, z punktu przecięcia linii składu destylatu z linią x = y wykreśla się linie q najmniej odchylone od linii x = y, biegnące w górę (ochłodzona ciecz) lub w dół (nagrzana para).
Po wykreśleniu linii q przeprowadza się linię operacyjną dolną. Jest to prosta łącząca punkty przecięcia prostej x = y z linią q i z linią składu cieczy wyczerpanej.
Ostatnim etapem procedury jest wykreślenie krzywej schodkowej między liniami operacyjnymi i linią równowagi fazowej, rozpoczynając od punktu przecięcia prostej x = y z linią składu destylatu. Liczba uzyskanych stopni określa liczbę półek teoretycznych w badanej kolumnie.
(1) W czasie destylacji roztworu A+B określa się ułamki molowe B w surówce (S), destylacie i cieczy wyczerpanej (W) oraz wielkość molowego strumienia (np. moli/h) destylatu i orosienia (R).

(2) Jeżeli dane, dotyczące termodynamicznej równowagi ciecz-para w układzie A+B, nie są w piśmiennictwie, muszą być wykonywane pomiary stężenia B w cieczy i parze w stanie równowagi.

(3) Dane dotyczące równowagi ciecz-para przedstawia się w układzie współrzędnych xB - yB:
(4) Wykreśla się linie określające skład surówki, destylatu i cieczy wyczerpanej:

METODA McCABE'A I THIELEGO KROK PO KROKU:
Górną linię operacyjną wykreśla się pod kątem zależnym od wielkości orosienia. "Linię q" wykreśla się pod kątem zależnym od stanu surówki.
|
Dolna linia operacyjna łączy dwa już wyznaczone punkty. Liczba "schodków" na linii prowadzonej między liniami operacyjnymi i linią rónowagi odpowiada liczbie półek teoretycznych.
Te same stężenia A i B w D i W można uzyskać w innych warunkach rektyfikacji.
7) Niedogon (niedogony, pogony, fuzel, olej fuzlowy, niem. fusel) - jest to produkt uboczny fermentacji alkoholowej, zbierany w końcowych etapach destylacji zacieru pdoczas produkcji spirytusu surowego. Jest to mieszanina szkodliwych produktów ubocznych fermentacji, takich jak alkohol pentylowy, izobutylowy, propylowy oraz z estrów niższych kwasów tłuszczowych. Powstawaniu fuzli sprzyjają źle dobrane warunki fermentacji (wysoka temperatura, niskie pH, niedobór azotu). Ma przykry zapach oraz smak. Występuje w samogonie (bimber, księżycówka) i spirytusie surowym. Niewielkie ilości niedogenów, i przedgonów (frakcji lżejszych od alkoholu etylowego) występują także w szlechetnych napojach alkoholowych produkowanych tradycyjnymi metodami destylacji - brandy, koniak, grappa, whisky. W trakcie leżakowania i dojrzewania ulegają korzystnym przemianom wpływającym na charakterystyczny smak trunków. Zazwyczaj w spirytusie surowym występuje koło 0,4% niedogonu. Usuwany jest na drodze rektyfikacji.
 |
| Źródło: pl.alcheek.com. |
 |
| Źródło: moonshine_still.republika.pl |
8) Orosienie (flegma, refluks, powrót, odciek) - jest to część pary opuszczającej kolumnę rektyfikacyjną, skroplona w deflegmatorze i zawracana na górę kolumny - pozostała część pary, po przejściu przez skraplacz, jest odbierana jako destylat (rektyfikat). Orosienie, spływając w dół w przeciwprądzie do pary, wzbogaca ją w składnik bardziej lotny. W niektórych procesach rektyfikacyjnych do orosienia dodaje się tak zwanej cieczy surowej.
1 - kolumna rektyfikacyjna, 2 - podgrzewacz, 3 - skraplacz, 4 - doprowadzenie surówki, 5 - wylot gazów, 6 - zawrót (orosienie), 7 - odprowadzenie destylatu, 8 - ciecz wyczerpana.
 |
| Źródło: WIkipedia. Schemat działania przykładowego reboilera kotłowego ogrzewanego parą. |
9) Reboiler - jest to rodzaj wymiennika ciepła służący do odparowania substancji ciekłej kosztem ciepła przekazanego jej od innego płynu (cieczy, gazu, pary wodnej). Reboilery są częścią składową przemysłowych systemów destylacyjnych. Umieszczone są zwykle u dołu kolumny rektyfikacyjnej i zasilane orosieniem. Generowana w nich para kierowana jest ponownie do kolumne destylacyjnej, a frakcje wysokowrzące usuwane są w formie ciekłej. W typowej przemysłowej kolumnie destylacyjnej cała ilość par substancji biorących udział w separacji pochodzi z reboilera.
 |
| Źródło: Wikipedia. Aqua Distillata. |
10) Woda destylowana - woda pozbawiona metodą destylacji, soli mineralnych oraz większości innych substancji ją zanieczyszczających. Zawiera rozpuszczone gazy (głównie dwutlenek węgla, także tlen i azot) oraz zanieczyszczenia lotnymi substancjami organicznymi. Stosowana jest w akumulatorach (jako rozcieńczalnik elektrolitu), w żelazkach parowych, w analizie chemicznej, w lekarstwach oraz wszędzie tam, gdzie wymagana jest wysoka czystość roztworu. Niektóre zanieczyszczenia wody można usunąć dokładniej przez: utlenianie związków organicznych, usuwanie twardości na wymieniaczach jonowych (woda demineralizowana), odgazowanie termiczne (woda odgazowana), korygowanie składu przy odsalaniu, kilkukrotną destylacją z dodatkiem kolejno kwasu fosforowego i wodorotlenku potasu lub sodu.
11) Woda redestylowana (woda podwójnie destylowana, łac. aqua bidistillata) - jest to woda poddana dwukrotnie destylacji w celu usunięcia zanieczyszczeń pozostałych po pierwszym procesie. Jednak nawet tak oczyszczona woda zawiera pewną (bardzo niewielką) ilość rozpuszczonych związków chemicznych i jej twardość wynosi około 0,04 mval/dm3, co jednak nie przeszkadza w większości zastosowań. Woda redestylowana jest znacznie droższa od wody demineralizowanej (dejonizowanej), lecz w niektórych przypadkach musi być stosowana, gdyż ta druga może zawierać resztki substancji organicznych (np. endotoksyny). Wodę destylowaną używa się m.in. do wyrobu leków okulistycznych.
12) Zeotrop (mieszanina zeotropowa) - jest to taki układ ciecz-para, w którym skłąd cieklej mieszaniny (roztworu) dwóch lub więcej związków chemicznych jest zawsze inny niż skład pary nasyconej, powstającej z tej cieczy. Na wykresach fazowych prężność pary - skład (izotermy) i temperatura - skład (izobary), ilustrujący warunki równowagi termodynamicznej, nie występują punkty ekstremalne, charakterystyczne dla azeotropów dodatnich i ujemnych. Jest to konsekwencją występowania niewielkich odchyleń od prawa Raoulta, dotyczącego roztworów doskonałych. Wśród układów zeotropowych wyróżnia się: homozeotropy z nieograniczona mieszalnością w fazie ciekłej; z ograniczoną mieszalnością w fazie ciekłej, w których obszar współistnienia faz ciekłych nie łączy się z obszarem współistnienia cieczy i pary; heterozeotropy, w któ®ych obszar współistnienia faz ciekłych łączy się z obszarem współistnienia cieczy i pary.

Poruszaj się po cyfrowym świecie randek za pomocą szpiegowanie Tinder! Zdobądź wgląd w działania na platformie, promując odpowiedzialne interakcje online.
OdpowiedzUsuń